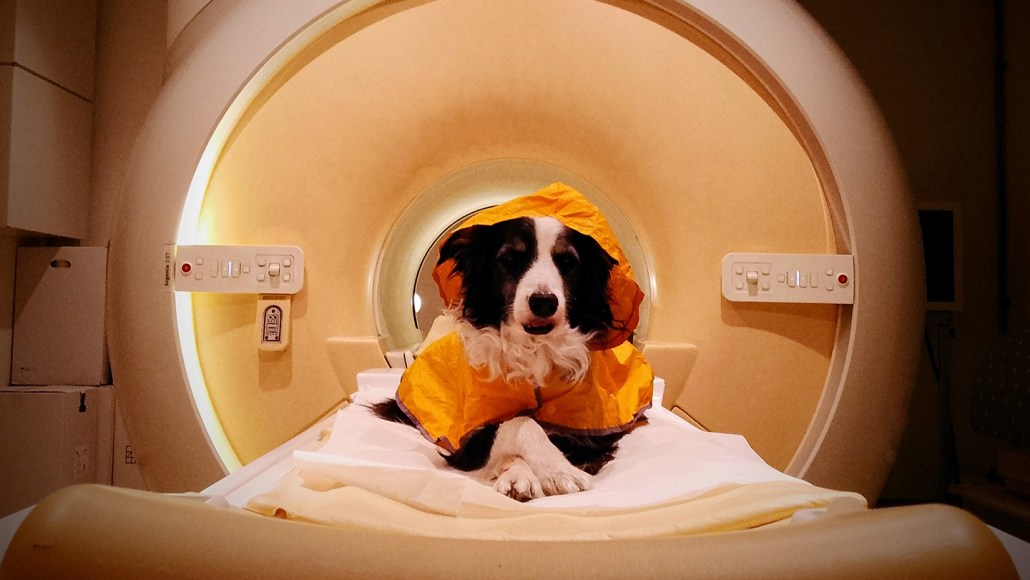
Kun-Kun the border collie underwent brain scans while looking at pictures of people’s faces. The experiments revealed that dog brains can differentiate between emotions.
LAURA V. CUAYA
Kun-Kun the border collie underwent brain scans while looking at pictures of people’s faces. The experiments revealed that dog brains can differentiate between emotions.
LAURA V. CUAYA