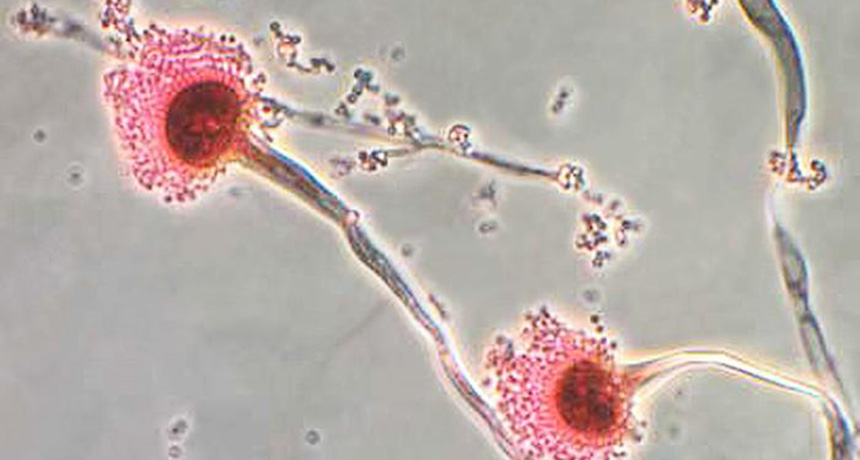
FUNGUS AMONG US Aspergillus fumigatus (shown) is a common fungus found in soil. New immunology research is helping explain why the organism can make immunocompromised people so sick, while going undetected by healthy people.
CDC
Immune cells can turn certain invaders on themselves, forcing them to prematurely self-destruct, researchers have discovered.
In mice, when white blood cells in the lungs engulf spores of a common airborne fungus, these immune cells release an enzyme that sends the fungal cells into programmed cell death. That prevents the spores from setting up shop in the lungs and sparking a potentially deadly lung infection, the researchers report in the Sept. 8 Science.
Found naturally in soil and decaying organic matter, the fungus, Aspergillus fumigatus, releases airborne spores that are found in small doses in the air people breathe every day. The finding may help explain why most people can regularly inhale the spores and not get sick. In people with weakened immune systems, though, this natural defense system doesn’t work. This research could eventually lead to better treatments for these patients.
Programmed cell death is a natural part of a cell’s life cycle — a way for organisms to break down old cells and make way for new ones. “Research in the last couple of decades has shown that microbes can exploit [cell death] pathways to cause disease,” says study coauthor Tobias Hohl, an infectious disease researcher at Memorial Sloan Kettering Cancer Center in New York City. But this study shows that the tables can be turned. “Not only can microbes exploit this in hosts, but host cells can exploit these pathways to instruct certain microbes to kill themselves.”
“The idea that the host triggers the mechanism of [programmed cell death] as a way of defending against infection is very cool,” says Borna Mehrad, a pulmonologist at the University of Florida College of Medicine in Gainesville who wasn’t part of the study.
Hohl and colleagues identified a gene in A. fumigatus that puts the brakes on programmed cell death. The gene, AfBIR1, shares an ancestor with the human gene survivin, which also regulates cell death.
When the researchers amped up the activity of AfBIR1 in a strain of the fungus, half the mice infected with the spores died during the eight-day study period. (Mice infected with unmodified spores were fine.) Cues that would normally send fungal cells to their death didn’t register, so the fungus was able to grow in the mice’s lungs.
In another experiment, the scientists gave mice a drug called S12, which took away AfBIR1’s brake effect. As a result, the mice were able to fight off the infection. “Those two findings suggested to us that this fungal [cell death] pathway really is critical,” Hohl says.
Hohl did this research with a special variety of A. fumigatus that changes color when its suicide instructions kick in. That advance allowed the researchers to make observations that weren’t possible before, Mehrad says.
For instance, Hohl and his colleagues noticed that fungal cells being engulfed by neutrophils, a type of white blood cell, appeared to be undergoing programmed cell death. That suggested that neutrophil activity might set off fungal programmed cell death.
Neutrophils release an enzyme called NADPH oxidase, and mice deficient in the enzyme weren’t as good at fending off the fungus, Hohl found. That makes sense with clinical data in humans too. People with a genetic mutation that causes a deficiency in NADPH oxidase are particularly at risk for developing an Aspergillus infection, Hohl says. People who have fewer neutrophils, due to chemotherapy or HIV infection, for instance, also make less of the enzyme and are less able to resist a fungal infection.
Survival rates vary, but the U.S. Centers for Disease Control and Prevention estimates that 41 percent of organ transplant recipients who contract aspergillosis die within a year. Seventy-five percent of stem cell transplant recipients with the infection die in that same time frame. Someday, a version of S12 that’s modified to work in humans might be able to boost these patients’ defenses against A. fumigatus infections, Hohl suggests.
In the future, he wants to see whether the same mechanisms extend to other fungal species too.