Gene therapies for sickle cell disease come with hope and challenges
Pediatrician Erica Esrick discusses existing sickle cell treatments and an ongoing clinical trial

Erica Esrick (far right) talks with Helen Obando, a patient in a gene therapy trial for sickle cell disease, and her family in 2019.
HILARY SWIFT/The New York Times
Today, it’s clear that our genes not only cause many diseases, but also hold potential cures. But that wasn’t always the case. It wasn’t until 1949 that scientists first found the molecular culprit of a disease — its roots in the genetic code. The disease was the blood disorder known as sickle cell disease, an inherited disorder that causes severe and debilitating pain. Now, nearly 75 years later, researchers are developing gene therapies to cure it.
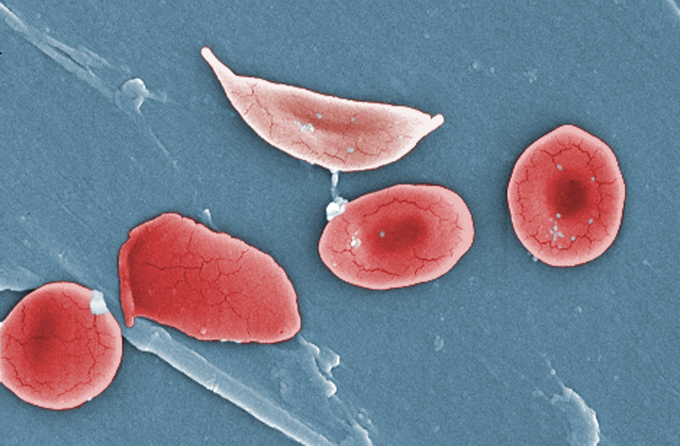
Sickle cell disease results from a change in a key protein in hemoglobin, which helps transport oxygen in red blood cells. Hemoglobin normally allows “red blood cells to be very floppy and pliable, and slip and slide through the blood vessels easily,” says pediatrician Erica Esrick. But a mutation in a single gene, the HBB gene, makes hemoglobin stack in long strings inside blood cells, giving them an inflexible, sickle shape. Instead of being “squishy,” the stiff red blood cells get stuck inside blood vessels, blocking blood flow.
Sickle cell affects millions of people around the world, particularly those whose ancestors come from sub-Saharan Africa, parts of the Middle East and Southeast Asia. In the United States, for instance, approximately 100,000 people live with the disease, most of them Black or Latino. People with sickle cell disease have a shortened life expectancy, living only into their late 40s on average, in large part due to strokes or organ damage from blocked blood vessels. Esrick, of Boston Children’s Hospital and Harvard Medical School, and others are trying to fight the disease through gene therapy.
To celebrate our 100th anniversary, we’re highlighting some of the biggest advances in science over the last century. To see more from the series, visit Century of Science.
Gene therapies seek to manipulate the very information of life by replacing, inactivating or fixing missing or broken genes — and so curing patients. But the journey to today’s handful of approved gene therapies, including for diseases like severe combined immunodeficiency syndrome, or SCID, certain blood cancers and spinal muscular atrophy, has been rocky. Early clinical trials in the 1990s weren’t effective, and the 2000s brought unintended and sometimes deadly consequences, including a leukemia-like illness.
Despite gene therapy’s challenges, many researchers believe sickle cell is a good target because the molecular pathways are well understood and straightforward. What’s more, every copy of the gene doesn’t need to be mended to have an effect. (Individuals who inherit the mutated gene from only one parent, for example, don’t develop sickle cell disease.)
Esrick is co-leading a clinical trial testing a gene therapy that attempts to encourage the body to make more of a healthy type of hemoglobin produced by fetuses and young babies — but not adults — called fetal hemoglobin. DNA for making a short string of genetic material called a microRNA is delivered by a virus into cells from a patient’s bone marrow. The virus, called a vector, permanently inserts the DNA into the cell’s genetic blueprint. The microRNA then interferes with the production of a protein that prevents fetal hemoglobin from being made. Once that protein is blocked, fetal hemoglobin production turns back on. Like turning on a faucet, a steady stream of the healthy hemoglobin can flow into the bloodstream, making up for the faulty form.
Preliminary data released in January 2021 showed that the treatment helped six sickle cell patients make fetal hemoglobin, Esrick and colleagues reported in the New England Journal of Medicine. During the follow-up period, ranging from several months to more than two years, the patients’ symptoms were reduced or eliminated. The team has expanded the trial to include more patients and further test the treatment.

Scientists are testing other ways to tackle sickle cell via gene therapy, too. A biotechnology company called bluebird bio is testing an approach that delivers a functional copy of the HBB gene to patients. Another team is preparing to begin a trial that will edit that gene directly using CRISPR/Cas9.
Science News staff writer Erin Garcia de Jesús spoke with Esrick about the ongoing fetal hemoglobin clinical trial, including the hurdles and the hope. The conversation has been edited for length and clarity.
Garcia de Jesús: What tools do we currently have to treat sickle cell?
Esrick: The only curative treatment is a bone marrow transplant. The bone marrow is like the factory for the blood cells. If you can get bone marrow from somebody who doesn’t have sickle cell disease, then you can grow your own healthy red blood cells that don’t sickle. But that is a major procedure, and it’s really only standard if you have what’s called a matched sibling [a brother or sister without sickle cell whose key white blood cell proteins match yours].
Less than 20 percent of people with sickle cell have a matched sibling available. If a matched sibling is available, then that’s a really good potential treatment option, but it is still a risky procedure. It comes along with some up-front risk of mortality and a lot of potential side effects, such as graft-versus-host disease and a higher risk of infection because of immunosuppressive drugs.
Then there are medications to treat sickle cell. The most well-established and long-lasting is called hydroxyurea. It increases fetal hemoglobin. In many people, it increases the fetal hemoglobin by a lot; that’s why it works so well. It’s been available since the ’90s, and has been moving gradually to younger and younger ages.
Now it is a very clear recommendation that essentially every child with sickle cell should be on it. But not everyone has access to specialized hematology care, and it’s a medication that has to be taken daily. Some people have adverse effects and can’t take it. It also doesn’t work for everybody.
Garcia de Jesús: How many people are in your team’s trial and what results have you seen so far?
Esrick: Nine patients have been treated. We anticipate the 10th patient will be treated soon. The preliminary data from the first six patients was published about a year ago. Additional data from subsequent patients has been largely quite similar — except for one patient whose fetal hemoglobin response was unfortunately not as robust.
Garcia de Jesús: What is the process like for the trial participants?
Esrick: Patients have to get their cells collected [the cells live in the bone marrow and give rise to blood cells], which takes a three-day hospital admission and sometimes has to be repeated a few times. It’s through IV, basically. Then the cells get taken off to the lab.
When we get word from the lab, “OK, we have a good product” [meaning the virus got the DNA into enough cells], then the patient comes back and is admitted to the hospital for a month or so. It’s a long and arduous hospital admission because they need to receive chemotherapy.
The reason they need chemotherapy is because the bone marrow cells that haven’t been collected need to get nearly wiped out in order to give the advantage to the cells that are being given back [also through IV] to set up shop and produce.
Chemotherapy comes with a lot of the side effects and risks associated with gene therapy, including acute short-term risks like hearing loss and nausea. And it also comes with some of the long-term risks, including infertility and a risk of blood cancers.
Garcia de Jesús: Why choose gene therapy over a bone marrow transplant if both require chemotherapy?
Esrick: With gene therapy, there’s no issue with immunosuppression, because it’s your own cells. People who get a transplant from another person have to be on immunosuppressive medications for a period of months after the transplant. There’s a risk of graft rejection because of the mismatch between the donor and the recipient.
The other risk in a bone marrow transplant from another person is graft-versus-host disease, where the graft and donated cells reject the recipient. That can cause severe disease. With gene therapy, that’s not a risk at all.
Garcia de Jesús: Last year, a clinical trial run by a company called bluebird bio announced that a trial participant developed leukemia. Cancer is obviously a huge concern and has thwarted previous gene therapy trials. What do we know so far about that?
Esrick: This was, of course, of major concern to the field. It was actually the second case of leukemia in that trial. The first one was published a couple of years ago as a case report.
If there’s ever a case of leukemia or any preleukemia in a gene therapy trial, we always ask: Was it caused because the vector stuck a gene into a spot that was dangerous?
It does not look like that’s the case. In the first patient in the bluebird bio trial who developed leukemia, the leukemia cells didn’t even have the transferred gene in them. So, the thought was that was probably just an example of chemotherapy causing leukemia, which we know can happen in a small percent of people who receive chemotherapy.
But the second case, in February 2021, really raised a red flag. Why is that happening two times in a trial of only 40-something patients? It’s still not exactly clear. There are some studies that suggest that people with sickle cell disease may have an increased risk of leukemia. But the [U.S. Food and Drug Administration] placed the bluebird bio trial on hold while some investigations were done. When it became pretty clear that it wasn’t directly related to the vector, the trial was allowed to reopen.
Our trial, which has many similarities to the bluebird bio trial, was not put on hold by the FDA but was put on hold by our funder, the National Heart, Lung and Blood Institute while they looked at the data. That hold was recently lifted.
We summarize the week's scientific breakthroughs every Thursday.
Garcia de Jesús: Have there been any cases of leukemia in your team’s trial?
Esrick: Fortunately, no.
But you know when anything like that happens in the field, it’s a big deal. I called all of the patients who we had treated in our trial to let them know. [The bluebird bio cases] happened in patients who had been treated three and five years prior. The longest-treated patient in our trial was almost three and a half years ago, and the most recently treated was about eight or nine months ago. I hope we see no concerning signs for any new development like that, but it’s too early to say.
Garcia de Jesús: What are some of the biggest challenges that sickle cell has had to overcome?
Esrick: For the longest time, there were no new therapies at all. These technologies took a long time because they are based on basic science discoveries that were being worked on. But also, the patient population with sickle cell is a population that has historically been underserved and without a lot of power.
In the United States, it’s primarily Black and Latino patients, and across the board those populations have suffered from health inequality. I think that if there were a disease that caused this degree of morbidity and mortality and pain in other parts of the population, it may have been speedier.
Garcia de Jesús: What gives you hope? What do you find exciting?
Esrick: I find myself bending over backwards to make sure that I’m not coming across as, “We have a cure!” But that said, it is really exciting that this is a treatment that is theoretically possible for everyone without needing to find a [bone marrow] match. That’s a huge difference from classic bone marrow transplants.
The speed at which new [gene therapy] treatments are being developed is amazing. I think the horizon is very bright in terms of one or maybe many of these therapies being really effective and safe. I’ve talked to so many patients and families who have reached out interested in our trial or other trials. There’s such a huge unmet need. The fact that there are a lot of these new treatments that are being developed is an encouragement to these families.