Sequencing the dead to save the living
Reviving ancient genomes of long-extinct creatures offers a window into past extinctions—and may help prevent future die outs
Genes tell stories of disease, of health, of parentage, all recorded in the chemical composition of DNA. But to many biologists, one of the most exciting tales that sequences of DNA letters can tell is an evolutionary one. And since evolution on its largest scale—the shifting cast of organisms populating Earth over the past few billion years—happens over lengthy periods of time, some of the best stories may be locked in the DNA of species long buried and gone extinct.
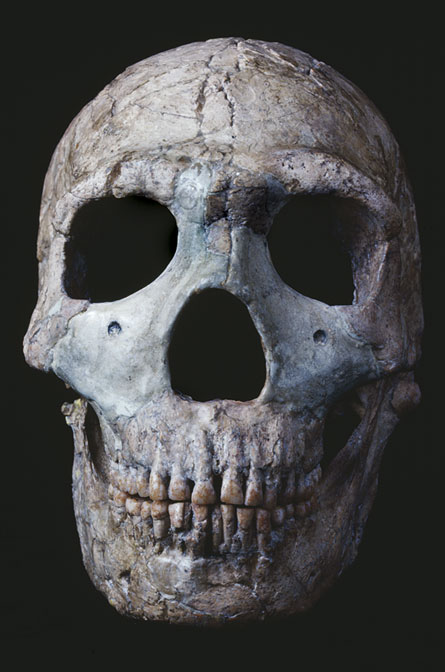
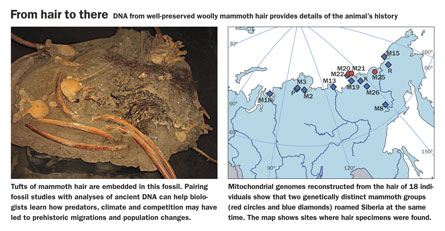

Sequencing the complete genome of a woolly mammoth that died 60,000 years ago or a Neandertal man who lived 40,000 years ago could reveal new details about prehistoric populations, their genetic diversity, how they changed through time and how they are related to species living today. It could also lead to deeper understandings of evolution’s details.
Biologists could use those genomes to study evolution almost as if it were a live performance, says ancient-DNA researcher Carles Lalueza-Fox of the Institute of Evolutionary Biology in Barcelona. Geneticists could pinpoint specific genes that helped mammoths and Neandertals survive, for a time. Experts might also identify the genes that left those species vulnerable to extinction.
Possessing the woolly mammoth or Neandertal genome couldn’t necessarily help experts resurrect the creatures Jurassic Park–style—at least not anytime soon—but the information could revise biologists’ views of the relationships between species extant and extinct. And the knowledge gained may help keep some of today’s threatened species, like Tasmanian devils, from disappearing, too.
Of course, “you can’t just pull the entire genome out of a cell that’s been dead for thousands of years,” says evolutionary geneticist Michael Hofreiter of the Max Planck Institute for Evolutionary Anthropology in Leipzig, Germany. The remains of extinct species sit in the ground for hundreds to thousands of years, and the genetic material decomposes, limiting how old genetic samples can be. Even in the best preserved specimens—a mammoth buried and frozen in the Siberian permafrost, for example—only short fragments of DNA remain intact and available for analysis.
In addition to decay, contamination of DNA in fossilized bone, hair and other ancient remains poses a major challenge. A fossil bone yields mostly DNA from microbes that ate away at the bodies of the dead animals. DNA from the organism of interest usually makes up less than 10 percent of the total genetic material extracted. Human DNA from handling specimens can also contaminate a sample—a factor particularly important in the sequencing of Neandertal DNA.
Still, in the past decade scientists have made enormous progress in extracting and working with ancient DNA. The shorter, more abundant mitochondrial genomes from many extinct species—Neandertals, mammoths, cave bears and the enormous Haast’s eagle, for example—have already been successfully sequenced.
And now genomicists are tackling the richer information stored in nuclear DNA. Experts are stitching together existing DNA fragments, repairing damaged ones and reconstructing missing ones. In November, scientists announced that they had completed sequencing about 70 percent of the woolly mammoth genome. If all goes well, the Neandertal genome will be finished up in the next few months.
Letter by letter
Decoding ancient nuclear DNA requires extracting as much material as possible from a specimen and then preparing the DNA so that a sequencing machine can read the chemical letters, or base pairs, that make up the code. Because the DNA is so degraded, it’s in fragments. “We are stuck sequencing tiny bit by tiny bit,” says Tom Gilbert of the University of Copenhagen. “In some of the mammoth hair samples, for example, the average read lengths are under 100 base pairs.” Sequencing the 4 billion to 5 billion letters that make up the mammoth genome in less than 100 base pair bits, Gilbert says, “would have taken a very, very long time.”
But that was before the development of faster, high-volume gene sequencers. These newer technologies, designed to deal with small stretches of DNA, can sequence hundreds of thousands of short fragments in one go. The additional speed also allows fragments to be resequenced several times over, which makes the results more reliable, says Hofreiter.
Fitting together the fragments into a complete genome comes next. Hofreiter says it’s like piecing together a puzzle. Genomicists look for similarities between extinct species’ and living species’ genes. If a matching sequence is found, genes of the extinct animal are lined up with the living animal’s genetic map. Comparing available Neandertal fragments with a human or ape genome, for example, helps genomicists compile a genetic blueprint of the extinct species.
Using such sequences, experts can pinpoint periods in history where species suffered drops in genetic diversity. By linking periods of diminished genetic diversity with events in an animal’s environment, scientists might even be able to reconstruct the history of the species.
Sections of the human genome, for example, hint that the genetic diversity of Homo sapiens declined between 150,000 and 50,000 years ago. Pairing genetic data with climate data from ice core samples, scientists have suggested that the number of breeding individuals fell to a few thousand, a drop possibly linked to a large volcanic eruption. But the analysis focused on just certain, small sections of the genome. Given that, as well as the difficulty in estimating the population sizes of early humans, any conclusion needs to wait for more genome studies, says genomicist Richard Green, who heads a team sequencing the Neandertal genome.
Green, also of the Max Planck Institute for Evolutionary Anthropology, thinks that by spelling out the complete Neandertal genome, for example, experts will discover points in time when individual genes in humans diverged from the genes in their extinct relatives. In the future, the sequences could reveal genetic changes that may have helped H. sapiens out-survive the Neandertals, he says.
Speech, skin and resurrection
Right now, geneticists focus on the ancient DNA fragments and genes that have been sequenced so far. One is FOXP2, a gene related to speech. The gene now appears to have the same sequence in humans and Neandertals. To study this gene in greater detail, a team of scientists inserted human FOXP2 genes into mice.
The mice didn’t talk, but they did seem to squeak differently from their nonmutant cousins, says Julia Fischer, who studies speech evolution at the German PrimateCenter in Göttingen. Speech is complex and involves many genes, but she says the work is a first step to knowing if Neandertals spoke to each other.
The next steps require geneticists to identify more human genes involved in speech and then scour Neandertal DNA to determine if it ever possessed those genes or variations of them. If those genes exist in Neandertal DNA, genomicists could extract or artificially reconstruct them.
If all these steps proceed as planned and the genes are inserted into mice, the animals still wouldn’t talk, but the findings might hint at whether Neandertals did, Fischer says. The experiment might also determine if language, and the cognitive ability to process it, was a factor that helped humans outlast the Neandertals.
Isolating more of the Neandertals’ supposed speech genes is still a few years off. But other ancient genes have already been brought back to life. In the past two years, geneticists have resurrected genes related to pigmentation from Neandertals and mammoths, for example.
In humans, the gene, called melanocortin 1 receptor or MC1R, codes for fair skin and red hair. When fragments of the ancient MC1R gene variants from Neandertals and mammoths were inserted into cells growing in the lab, the cells produced proteins from the fragments. Analysis of the proteins suggested that some mammoths might have been blonds, while some Neandertals might have been redheads, Lalueza-Fox says.
These experiments are the second closest that scientists have gotten to bringing back extinct species, he says. The closest effort came in May when genomicists reported resurrecting a fragment of DNA from a thylacine, or Tasmanian tiger. The carnivorous marsupial was not a true tiger but was given the name because of its stripes. A cousin of the Tasmanian devil, the tiger competed with the devils for food before disappearing into extinction in the 1930s.
To see if the thylacine’s DNA would still function, scientists extracted a gene linked to collagen production from an alcohol-preserved specimen. They then inserted the gene into mice and showed that it switched on a marker gene in cartilage-producing cells of the mutant mice’s embryos. That demonstrated, for the first time, that old, preserved DNA can be brought back to life—at least in mice.
But resurrecting one gene is much different from resurrecting the thousands or tens of thousands of genes that make up an animal’s genome, Lalueza-Fox says. Bringing back a thylacine, mammoth or Neandertal is decades away, he says. It’s also not on many scientists’ agendas. Using genome-sequencing technology to save the Tasmanian devil from extinction, however, is.
Devil of a problem
“Some people say, let Tasmanian devils go extinct, then sequence them. But it’s more warranted to do it beforehand when we can save them,” says genomicist Stephan Schuster of PennsylvaniaStateUniversity in University Park. “That’s why we’ve crossed over from sequencing the dead to the living.”
He and Penn State colleague Webb Miller have been working to sequence fragments of the woolly mammoth genome and, reporting in the Nov. 20 Nature, just released a sequence covering 70 percent of the nuclear genome.
That work helped them design the software for sequencing the Tasmanian devil genome. But the two scientists soon discovered they didn’t have as detailed directions for spelling out the devil genome as they did for mammoth DNA.
The Tasmanian devil’s closest living relative is the South American opossum, Miller notes. Sequenced DNA from both species shows that only 90 percent of their genomes overlap. Elephants and mammoths share some 99.4 percent of their DNA, Miller says. That’s enough overlap for experts to understand the role of mammoths’ noncoding regions, formerly dubbed “junk DNA.” It’s a lot harder to understand the importance of the devil’s noncoding regions because of the greater number of gaps in the code.
“We need to know what’s going on in those pieces of devil DNA, though, because a lot of the guys are catching cancer,” Miller says.
Cancer is ordinarily not contagious. But a cancer that affects Tasmanian devils is spread by biting, aggressive mating or eating infected food. The devils’ small island habitat means that the cancer spreads more quickly. Of the adult Tasmanian devils that conservationists have trapped and tested, 83 percent show symptoms of the disease. It appears first as lesions and lumps around the mouth and can spread from the face to the entire body. With time, the facial tumors grow so big that a devil can’t eat and starves to death.
To better understand the facial tumor disease, Miller and Schuster are sequencing and comparing the genomes of two devils—Cedric and his half brother. Cedric is a star of the Tasmanian media because he is resistant to the cancer. If another devil with the contagious cancer bites him, his body can tell his cells from the dangerous cells, and the cancer doesn’t take hold. The devil’s half brother isn’t so lucky. He’s not resistant.
Finding major differences between Cedric’s and his brother’s genes would be “almost too cool,” Miller says. The genetic variations would provide clues about how the cancer interacts with the creature’s genes. Using that information, genomicists could identify which devils are fully or at least partially cancer resistant. “It’s conceivable that we might be able to say, here is this variation in the devils’ genes. You have this version, you’re toast,” Miller says. “And you, you have the other version, you might live.”