Tumor Tell-All
Unraveling complex genetic stories in cancer cells may lead to personalized treatment
Tumors are ugly. But the staff at Massachusetts General Hospital takes a snapshot of almost every one that crosses the doorstep.

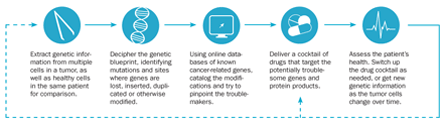
Unraveling complex genetic stories in cancer cells may lead to personalized treatment
Tumors are ugly. But the staff at Massachusetts General Hospital takes a snapshot of almost every one that crosses the doorstep.