Computer compatibility tests might help flu-fighting drugs find their groove.
A pandemic of the H1N1 swine flu virus has health officials worried that the virus could develop resistance to drugs such as Tamiflu used to treat infected people. A new computerized screening method could help find new or already existing drugs that find a flu virus’ weak spot, researchers from the University of California, San Diego reported December 6 at the annual meeting of the American Society for Cell Biology.
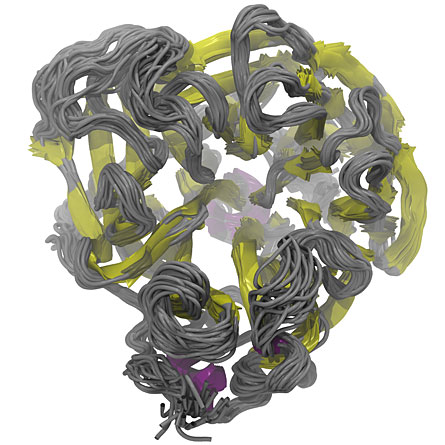
Researchers Daniel Dadon, Jacob Durrant and J. Andrew McCammon, all of UCSD, made a computer movie of slight structural shifts occurring in the neuraminidase 1 enzyme (the N1 in H1N1 and H5N1), a protein found in the avian and swine influenza viruses. Those changes reveal possible target areas that could allow drugs to circumvent a virus’ usual means of becoming resistant.
All influenza viruses have a neuraminidase enzyme, but the protein comes in several subtypes. Previous work had shown that the N1 subtype contains a loop that makes it more flexible than other neuraminidase subtypes, says Rommie Amaro, a computational biologist at the University of California, Irvine. “It is particularly nimble,” she says. The enzyme’s flexibility could affect the way drugs bind to it.
Analyses of still frames from the simulation, which is called a relaxed complex scheme, revealed 27 different natural conformations that the N1 protein could take on under conditions it might encounter in a host cell. Some parts of the protein change shape readily and some stiffer portions are locked into place, the researchers discovered.
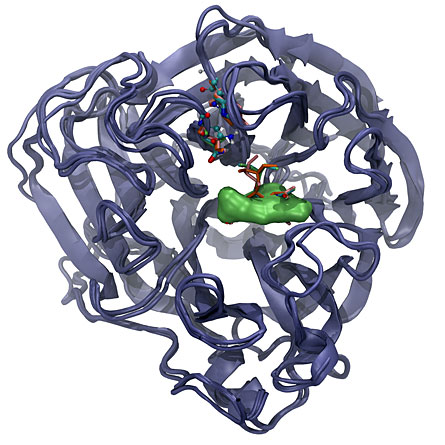
Drugs currently used against flu — including oseltamivir, better known as Tamiflu, peramivir and zanamivir — all insert themselves into neuraminidase at about the same location within that enzyme. When the drugs insert into that pocket, they block the enzyme’s ability to release newly made viral particles from the cell, and this blockage prevents the spread of the disease.
That location is prone to structural changes such as those revealed by the simulation, and to genetic changes that affect the amino acid building blocks that compose the protein. Many of those amino acid changes also alter the shape of the pocket, keeping the drugs from binding and thus making the flu virus resistant to the drugs.
To find drugs that could block the protein’s active site in a different way — and knock out viruses resistant to the currently used drugs — the researchers mined a library of FDA-approved drugs. The team digitally sliced up the drugs and simulated how the drug fragments might bind to all of the enzyme’s forms.
Among those fragments, the team found 15 novel compounds that could wedge into the protein’s pocket and block its action better than Tamiflu or other antiviral drugs would. A closer examination revealed that those 15 compounds share a common structure. What’s more, the compounds lodge into a part of the protein that doesn’t allow changes easily, meaning that those areas are less likely to mutate and develop drug resistance than the parts of the protein that come into contact with Tamiflu and other current flu treatments, Dadon says.
But because the computer-generated fragment molecules don’t exist in the real world, the researchers needed to see whether any existing, small molecules could work just as well. Searching four databases of drugs turned up six small molecules that had the same common structure as the digitally diced compounds. These real compounds are currently being tested by collaborators in Australia to determine whether they really do block the flu’s action.
Both of the approaches Dadon’s team followed in the new drug design scheme — examining all forms of the protein and then screening a library of fragments from approved drugs — could be easily adapted for other molecules, Amaro says. The caveat is that researchers need to have prior knowledge of an enzyme’s structure in order to develop effective drugs, she says.