Attacking Alzheimer’s
Comprehending the causes gets more complex
It starts simply enough, with those memory failures that are common to everyone: losing keys, getting lost in an unfamiliar place, or forgetting the name of a new acquaintance. As months and years go by, however, people suffering from Alzheimer’s disease have trouble recognizing where they are, have difficulty writing and speaking, and forget how to perform routine daily tasks like washing their hands or getting dressed. Eventually, all sense of self seems to vanish, and the person becomes completely dependent on others for care.
Alzheimer’s affects up to 4 million people in the United States, with about 360,000 people developing the disease each year. The number of new cases will probably rise as the population ages. Although Alzheimer’s becomes more common as people grow older, but it’s not a normal part of aging (SN: 3/10/01, p.156: Making Sense of Centenarians).
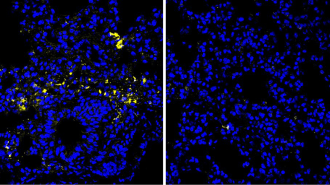
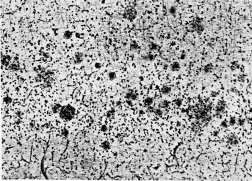
Alois Alzheimer first described the disease in 1907 after he examined the brains of people who had died of a severe dementia. The German psychiatrist noted the presence of abnormal protein deposits, or plaques, piled around nerve cells in the brain and tangled proteins inside the cells. These so-called beta-amyloid plaques and tau tangles have come to be considered the signature of Alzheimer’s disease. Researchers have linked these plaques and tangles, which develop first in the area of the brain used for memory, to the decline in connections between nerve cells and the death of brain cells–changes presumed to be responsible for the brain dysfunctions of the disease.
In the past few years, most scientists studying Alzheimer’s have considered the beta-amyloid plaques to be the crucial factor underlying Alzheimer’s disease. Yet some researchers now suggest that this so-called amyloid hypothesis is overstated and that other entities, including tau tangles, are as important as beta-amyloid.
Simplest explanation
“The amyloid hypothesis is the simplest explanation possible,” says Steve Estus of the University of Kentucky in Lexington. A variety of research, ranging from studies of cell cultures to the genetics of the disease, support it. Estus says that opponents of the amyloid hypothesis aren’t seeing the whole picture provided by that evidence.
Beta-amyloid is a tiny protein fragment snipped from a larger protein called amyloid precursor protein. Plaques are a mix of beta-amyloid with remnants of nerve cells and other brain cells.
Tests of nerve cells grown in lab cultures suggest that, in some forms, beta-amyloid itself can be toxic, producing compounds called free radicals that damage nerve cells. Moreover, beta-amyloid plaques are associated with swelling and inflammation, a process that might signal the early stages of nerve cell death.
What’s more, Estus says, the genetic mutations associated with rare inherited forms of Alzheimer’s disease seem to increase the concentration of beta-amyloid deposits in the brain, and this increase is typically seen before symptoms develop.
Several animal models mimic different aspects of Alzheimer’s disease, though scientists disagree about how applicable any one model is to people. Mice genetically engineered to produce higher-than-normal amounts of human beta-amyloid or the amyloid precursor protein can develop beta-amyloid plaques and, in some cases, show memory impairment, such as difficulty navigating a maze.
What many researchers call the true test of the idea that beta-amyloid deposits underlie Alzheimer’s disease is now under way. It asks whether a vaccine against beta-amyloid reduces the illness’ characteristic symptoms. Immunizing mice against the protein fragment reduces the number of plaques in their brains and seems to slightly boost cognitive function (SN: 7/15/00, p. 38: Possible Alzheimer’s vaccine seems safe). Small studies of a vaccine against beta-amyloid indicate that it’s safe for people, and researchers are now testing whether it wards off Alzheimer’s in people whose family histories put them at high risk of the disease.
If the vaccine does benefit the people taking it, it will be clear-cut confirmation that beta-amyloid deposition in the brain is one of the key factors pushing people toward Alzheimer’s, says David Morgan of the University of South Florida in Tampa, who is studying a vaccine in mice.
The vaccine trial results will be important, agrees Stephen R. Robinson of Monash University in Clayton, Australia. However, he doesn’t expect a positive result because the bulk of the research to date doesn’t prove that beta-amyloid causes Alzheimer’s, he argues.
“If one has a loose-fitting shoe, one will often see on one’s heel a blister,” he cautions. “This doesn’t mean the blister caused the loose-fitting shoe.”
The small group of researchers challenging beta-amyloid buildup in brain plaques as the primary cause of Alzheimer’s is becoming increasingly vocal. Their views were highlighted in a July meeting in Cincinnati called Challenging Views of Alzheimer’s Disease.
The idea that beta-amyloid causes Alzheimer’s symptoms is “far and away the most popular hypothesis,” says John Blass of Cornell University-Weill Medical College in New York. “But truth lies in the eye of the beholder. Looking at the same data, you can either see the doughnut or you can see what to some of us looks like a very big hole.”
Blass and like-minded scientists point out that the progressive mental deterioration marking Alzheimer’s disease and the appearance of beta-amyloid plaques don’t necessarily go hand in hand. As many as 20 percent of people diagnosed with Alzheimer’s show no sign of beta-amyloid plaques at autopsy. Likewise, up to 50 percent of people who have relatively high numbers of beta-amyloid plaques in their brains at autopsy showed no sign of mental impairment during their lives.
Proponents of beta-amyloid’s causative role in Alzheimer’s often explain these findings in two parts. First, people without plaques probably have a form of dementia that isn’t actually Alzheimer’s. Second, people with beta-amyloid deposits but no Alzheimer’s symptoms probably started with brain reserves. These people might started with brains that are bigger than average, or they might have more connections between nerve cells than do people who succumb to Alzheimer’s.
Symptom, not cause?
Many of the mouse strains developed to mimic Alzheimer’s disease don’t show signs consistent with the amyloid hypothesis, Robinson says. Some of these mice have higher concentrations of beta-amyloid in their brains than Alzheimer’s patients do and yet are significantly less likely than those patients to show a drop in brain function.
In the June 15 Journal of Neuroscience, a team of researchers reported that mice engineered to rapidly produce beta-amyloid plaques show signs of nerve cell damage even before the plaques appear. By 8 months, the engineered mice showed more signs of brain damage from free radicals than did nonengineered mice, and by a year, the beta-amyloid mice had about twice as much free radical damage. Yet beta-amyloid plaques were still undetectable in the engineered animals at 8 months of age. A surge in brain concentrations of beta-amyloid wasn’t detectable until the engineered animals were about a year old.
“Our research indicates that [free radical damage] is probably a primary event in the course of the illness,” says Domenico Pratico of the University of Pennsylvania School of Medicine in Philadelphia. Since free radical damage occurs before beta-amyloid buildup, the study suggests that plaque formation isn’t the trigger for Alzheimer’s disease.
Robinson notes that many of the genes that have mutations linked to Alzheimer’s disease are also involved in normal cell regeneration. These genes’ proteins, which are responsible for repairing stress-related damage, become less efficient as people age. In cell cultures, some forms of beta-amyloid also seem to protect cells against stress and damage, Robinson says.
The cell damage and beta-amyloid are both in the brain, says Mark A. Smith of Case Western Reserve University in Cleveland. But it doesn’t prove that beta-amyloid is a bad guy, he argues. Beta-amyloid may appear to cause damage while it’s actually offering a defense–as a firefighter spraying water damages a building while trying to save it.
“Imagine if beta-amyloid is a fireman,” he says. “It may be destructive, but it may be a key component of a protective response.”
If the doubters of the amyloid hypothesis are correct, the beta-amyloid vaccine now being tested might actually weaken the brain’s response to damage. So far, however, studies in mice and in people give no evidence of that.
Another possibility
If beta-amyloid isn’t the culprit in Alzheimer’s disease, what is? One possibility, still being aggressively pursued by many researchers, is that the tau tangles–the second hallmark of Alzheimer’s–hold the key to understanding Alzheimer’s disease.
Normally, tau proteins serve as the ties holding together microtubules, which can be thought of as railroad tracks along which nutrients and proteins move in a cell. In Alzheimer’s disease, these tau proteins often have extra phosphorus molecules tacked on. Instead of stabilizing the microtubule tracks, these phosphorylated tau proteins tangle and derail the system. The collapse of this transport system may impede communication between nerve cells and lead to their death.
By looking at mutations, brain researchers have linked abnormalities in tau proteins to other forms of dementia. However, no mutation in any of the genes that produce tau proteins has been linked to Alzheimer’s disease. Some researchers are examining whether the presence of phosphorylated tau proteins, rather than the tangles themselves, causes the cell damage and dysfunction seen in Alzheimer’s.
Rather than working individually, beta-amyloid plaques and tau tangles may act in concert. To create one of the newest animal models of Alzheimer’s disease, Michael Hutton at the Mayo Clinic in Jacksonville, Fla., has interbred mice with mutated human genes for the amyloid precursor protein and mice with mutated human tau genes. The offspring have more tangles and more beta-amyloid plaques than their parents do, especially in a brain area critical for memory, Hutton reports in the Aug. 24 Science.
In another attempt to assess plaques’ and tangles’ combined effect, Peter Davies of the Albert Einstein College of Medicine in New York has recently shown that a protein called Pin-1 binds to both the amyloid precursor protein and tau. In a test tube, Pin-1 can make phosphorylated tau proteins resume their normal function.
Pin-1 might therefore be a useful treatment for people with Alzheimer’s. But the protein which participates in normal cell division, so any extra may increase formation of amyloid precursor protein and have a deleterious effect on a person’s brain, says Davies. Though researchers are still teasing out Pin-1’s effects, the protein may link the two major features of Alzheimer’s disease pathology.
Cell-division proteins
An emerging field that may shed additional light on the causes of Alzheimer’s is the study of proteins that regulate cell division. Such proteins aren’t normally present in many nerve cells in the adult brain because those cells don’t typically divide. Over the past year, however, studies have identified Pin-1 and several other cell-division proteins in the brains of people with Alzheimer’s or of mice with Alzheimer’s-like symptoms.
At the Cincinnati meeting, participants divided over how to interpret these results. About half held that the cell-division proteins are present simply because they play a role when damaged cells try to repair themselves. The other researchers argued that the cell-division proteins lead the cell to its death.
Once the specialized brain cells are prompted to start dividing, they’re marked for destruction, argues Thomas Arendt of the University of Leipzig in Germany.
According to yet another new view, brain cells run low on fuel in people with the disease. Experimental conditions that reduce the brain’s metabolism in animals typically induce deficits in learning and memory, Blass points out. In Alzheimer’s patients, measures of brain metabolism typically drop before symptoms appear. Researchers measure metabolism in people who show small drops in cognitive function, according to sensitive tests, but continue to function normally. This group is at high risk for developing Alzheimer’s.
Lack of brain energy may explain the mental decline seen in Alzheimer’s patients, Blass says. The presence of plaques or tangles might be either a cause or effect of impaired metabolism, he says.
“The important point is, this [metabolic defect] may be a point for treatment,” Blass says.
At the Cincinnati meeting, he reported a study of seven Alzheimer’s patients given daily doses of a drink designed to improve brain metabolism. The drink contained sugar and a compound called malate, which boosts the function of mitochondria, the powerhouses of cells.
Over 3 months, each of the patients improved his or her mental functioning as measured on standard tests, Blass reports. He and his colleagues are now attempting a larger study in which some participants will get the metabolism boost and others will receive a placebo. During the study, neither the patients nor physicians will know which treatment each volunteer is given.
Researchers on both sides of the amyloid-hypothesis debate admit that the truth could lie in some middle ground, since Alzheimer’s is a complicated disease probably triggered by many factors.
“The science of Alzheimer’s is a work in progress,” says Estus, adding that most of the evidence so far supports beta-amyloid as the trigger for Alzheimer’s disease.
“The amyloid hypothesis has a lot of truth to it,” Blass says, “but data that don’t support it must still be respected [instead of] being shouted down.”