The Inconstant Gardener
Microglia, the same immune cells that help sculpt the developing brain, may do damage later in life

Composite: Fabler/iStockPhoto, dem10/istockphoto, egeeksen/iStockPhoto, adapted by S. Egts
Like the cavalry in old Western movies, certain immune cells in the brain rush to answer distress calls and save the day. If a nerve cell is injured or a toxin attacks the brain, these microglia ride to the rescue, moving to the injury site and destroying any bad guys they encounter.
But even in the movies the cavalry, mistakenly or intentionally, sometimes mows down innocent bystanders. A similar scenario may be playing out in the brain with processes that normally limit damage: Microglia may be improperly activated late in life and play a role in neurodegenerative disease.
Take Alzheimer’s as an example. People with Alzheimer’s disease lose millions of nerve cells in brain areas crucial for memory. As the losses grow, the brain also becomes cluttered with clumps of plaque containing a protein fragment called amyloid-beta, or A-beta. Where these plaques emerge, microglia appear as well.
For years, researchers have believed that the microglia are there to help clear out dead cells and debris left behind as the plaques progressively damage the brain. But new research suggests a different scenario. Long before the plaques collect, microglia may be called in and compelled to attack healthy cells.
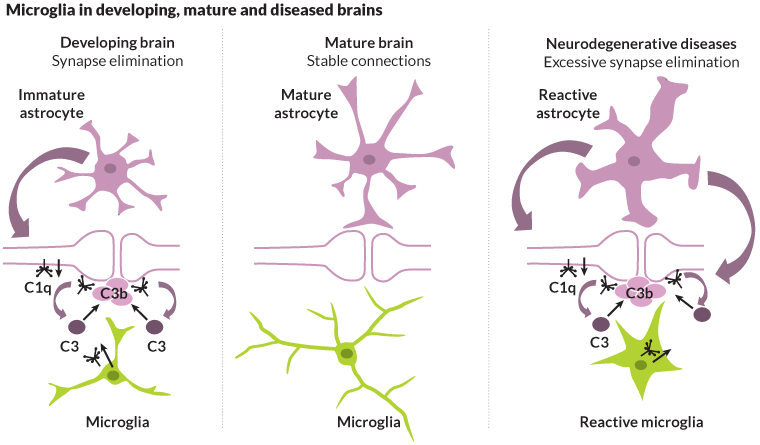
A protein called C1q may set up the brain for this destruction. C1q is found in abundance in young brains. During development, the protein works with microglia to target and prune unused connections between brain cells, making way for new connections to form in response to experience and learning.
C1q also accumulates in aging brains, according to recent research. The protein collects at synapses, the junctions between nerve cells where communication between the cells takes place. Information is processed and stored in the brain through these delicate connections. Each nerve cell can have thousands of synapses, creating a total of billions of connections inside the brain.
What impact does accumulating C1q — and its troops, the microglia — have on the older brain?
Stanford neuroscientist Ben Barres speculates that microglia may be reactivated later in life in response to a head injury, stroke or severe illness. This reactivation, he says, may explain the massive loss of neurons seen in neurodegenerative disease.
“Our findings suggest that long before the [nerve] cells die, their synapses are dying,” Barres says. “It may be that the neuron dies only after it has lost a sufficient number of synapses.”
That microglia have the potential to act as both protector and attacker doesn’t come as a complete surprise. But whether these immune cells are helpful or harmful in these diseases has remained unclear.
While pruning of synapses during development is necessary for wiring the nervous system, activation of microglia in old age can occur through various means and is a detrimental process, says R. Douglas Fields, chief of the nervous system development and plasticity section at the National Institute of Child Health and Human Development.
“This is somewhat similar to the situation where the action of the body’s immune system is normally crucial, but in autoimmune disorders such as multiple sclerosis, pathological consequences can result from the mechanism intended to protect the body,” Fields says.
By studying how microglia respond to various proteins and signals in the brain, in both health and disease, scientists aim to clarify microglia’s role in synapse loss and disease. Some researchers are also devising ways to tweak microglia’s function in the body by inhibiting immune responses that call microglia to action or replacing faulty microglia with healthy ones.
Guardian and housekeeper
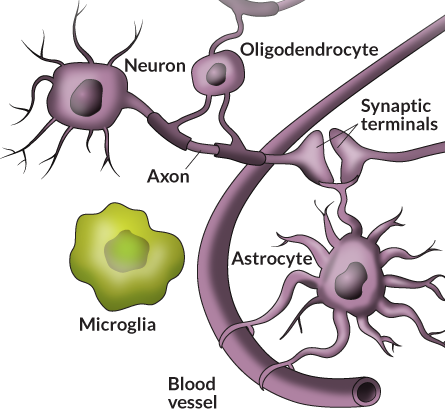
Microglia belong to a class of brain cells called glia — the name literally means glue — which also includes astrocytes and oligodendrocytes. Together, these cells outnumber neurons 3 to 1. While neurons do the heavy work of transmitting and processing information, glial cells produce proteins necessary for the health and survival of the neurons.
Astrocytes ferry nutrients to neurons and help control where synaptic connections form. They also promote neuron survival by stimulating the growth of axons, the long nerve fibers that transmit neuron impulses. Oligodendrocytes coat the axons with an insulating sheath called myelin. Because axons serve as nerve cells’ communication fibers, much like electrical wires, the insulating myelin coat helps to boost signal speeds.
Hard-working microglia serve as both guardian and housekeeper. If something bad happens in the nervous system — a blow to the head or a bacterial infection — these small cells take action. Pulling in their long, skinny appendages, they puff up in size and move toward the injury. Once there, they release chemical substances to kill invaders then gobble up any bits of debris, including dead or dying cells. By keeping the environment around the neurons and synapses clean, microglia help keep things running smoothly.
For decades, microglia were considered supporting players that remained “at rest” unless called on to respond to an emergency. As better imaging tools became available and allowed scientists to peer inside the brains of living animals, it became clear that, for these cells, there is no rest.
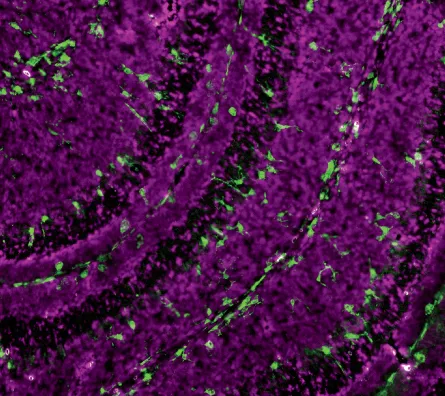
In 2007, Beth Stevens, a postdoctoral fellow in Barres’ lab, made an even more surprising observation while spying on brain cells of young mice. The microglia she saw were not only brimming with activity, they were already in their puffed-up fighter mode. Last year, Stevens, now a Harvard neuroscientist, confirmed that microglia help sculpt the brain’s circuits by pruning unneeded connections between neurons during early development, when the brain forms more neurons and synapses than it needs.
In the study, Stevens showed that microglia take their cues, in part, from a set of signals that trigger an immune response called the complement cascade. In the body, complement proteins bind to invading pathogens or dying cells, sparking their destruction by immune cells called macrophages. The multistep process involves about 20 different proteins, which one by one glom onto unwanted bits and debris, ultimately calling upon the macrophages to gobble up the invader.
In the brain, microglia do the work of macrophages but with an added job: pruning. Stevens’ group found one complement protein in particular, called C3, on synapses destined for pruning during early development. Microglia have proteins that lock onto C3, which acts as a biochemical bugler calling microglia to action.
Stevens’ group used dyes to track microglial activity in the visual system of mice and caught microglia in the act of pruning synapses tagged with C3. In the study, published last year in Neuron, Stevens noted that microglia engulfed synapses only during development, when extra synapses are forming and unused synapses need clearing away. Stevens’ lab is studying whether microglia play a role in maintaining the brain’s synaptic architecture in adult mice.
Coaxed into battle
In most cases, the complement system works for the brain’s benefit, calling on microglia to clear unwanted debris or unneeded connections. But if left unchecked, complement proteins can coax microglia into battle with the cells they should be protecting. That’s something that Barres believes happens in a number of neurodegenerative conditions.
He has some evidence to back this up. Barres’ group found that synapse elimination is reactivated very early in glaucoma, a neurodegenerative disease that is a major cause of blindness. In the 2007 study, his team showed that the earliest sign of the disease was the activation of the complement cascade at synapses. Soon after came massive synapse loss.
Synapse loss is a hallmark of a number of diseases, including Alzheimer’s. Pointing to recent findings that show C1q, a key complement protein, increasingly collects at the synapses with age, Barres says immune signaling may be at play in such diseases as well.
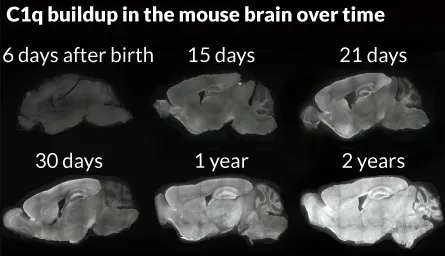
C1q is first in the series of proteins that initiate the complement cascade. Examinations of mouse and human brain tissue show as much as a 300-fold increase of the protein in aging brains, even healthy ones. Studies from other labs have shown the presence of both microglia and C1q in areas where A-beta plaques accumulate in patients’ brains.
“By the time you get to be a 2-year-old mouse or a 77-year-old human, the C1q is pretty much everywhere throughout the brain,” Barres says.
What’s more, the most dramatic increases are seen in the hippocampus and the substantia nigra, brain regions that are typically ravaged by Alzheimer’s and Parkinson’s diseases.
But just because C1q is present doesn’t mean microglia are going to come running. Unlike C1q seen in brain development, C1q found in the healthy, aging brain is not activated, a step needed to draw other complement proteins and microglia to the site.
What it does mean, Barres says, is that these delicate connections are “primed” with complement protein, poised to call out for help if injury or major illness strikes. Because various stresses lead to a full-blown immune response, a stroke, a blow to the head or even a bad case of pneumonia could put the tagged synapses on a path toward destruction by calling microglia into action.
“There’s always been this mystery: Why is the old brain selectively vulnerable to neurodegenerative disease?” Barres says. “I believe that this could be the missing link. It’s only the old brain that has already got the complement system essentially charged up and ready to go.”
Spreading destruction
Nerve cells are uniquely vulnerable to activation of the complement cascade. Other cells in the body have complement-inhibiting agents to prevent the loss of healthy tissue during an immune attack. Such fail-safe mechanisms allow the complement system to attack bacteria on the liver without harming the liver itself.
But neurons have little or no complement inhibitors. If the complement system gets out of control and microglia begin gobbling C1q-coded connections in an aged brain, according to Barres’ theory, a spreading “fire” may burn through the synapses, destroying neural circuits and the memories that go with them.
“You can imagine in the brain, where you have this dense network of synaptic connections, all jam-packed next to each other, if the complement system gets set off at one of those synapses, there will be innocent bystander killing,” he says.
This scenario has yet to be tested. But it could help explain the massive synapse loss seen in a host of neurodegenerative disorders. In the Aug. 14Journal of Neuroscience, Barres and his group report that mice genetically deficient in C1q age with fewer memory problems than normal mice.
Scientists are trying to figure out why C1q targets some synapses and not others, or what series of events puts C1q on the path of destruction. If researchers could find the key players involved, they may be able to develop new treatment approaches to prevent synapse loss, Barres says.
Even as they expand their studies to see what happens between C1q buildup and microglia activation in the brain, Barres and his colleagues are developing a drug to inhibit immune responses that call microglia to action in aging brains. Barres has created a start-up company, called Annexon, to develop drugs to block the action of C1q and other proteins in the complement cascade. One drug is already being tested in animals.
Fatigued fighters
Attacking healthy cells may not be the only way that microglia contribute to disease. Some scientists point to another possibility: With age, microglia may lose the oomph they need to counter an assault.
Microglia become activated in increasing numbers in aging brains, Fields says, which has long been considered a sign of the cells’ hard work to protect the brain from accumulating insults. It’s also thought to be a reason people recover from strokes better at younger ages.
“If the brain’s microglia are already activated chronically before a stroke, they cannot mount a response to the injury that is as aggressive as if they were quiescent in a younger brain,” he says.
The situation may be made worse by disease. Researchers led by Helmut Kettenmann of the Max Delbrück Center for Molecular Medicine in Berlin recorded moving images of microglia surrounding plaque deposits in the brains of mice with an Alzheimer’s-like condition using two-photon microscopy. The microglia could not perform normal custodial functions: They did not clear cell fragments or other structures from the brain and lost their ability to move about in their surroundings.
When the scientists injected the mice with an antibody against the A-beta protein, prompting the rodents to produce fewer plaques, the microglial cells resumed their normal functions.
How microglial cells become weakened in Alzheimer’s is not fully understood, Kettenmann says. His group is now studying the roles of various receptors in microglia. For example, different receptors on microglia may initiate tissue repair, inflammation or neurodegeneration. He’s also studying how microglia participate in other diseases, such as cancer. Four years ago, his group published findings that show microglial cells support the growth of some tumor cells, helping them spread through the brain.
“In the old days we thought that microglia activation worked like an action potential — an all or nothing process,” says Kettenmann. “Now, we recognize that the activation process is quite diverse,” depending in part on the condition that sets off the alarm.
As scientists explore the workings of microglia in disease, other efforts aim to see how microglia perform in normal, healthy brains.
“We know almost nothing about their functions in a normal brain,” Kettenmann says. “It’s likely that many of the mechanisms which are used in pathology are also used in development and plasticity.”
This seems to be the case in other kinds of cells, he says. Rather than inventing a new path for destruction, cells take an untimely misstep on pathways meant for normal development or growth.
“Somehow the microglial cell may get put back on that pathway,” Kettenmann says. And maybe there’s a way to set it right again.
Do microglia fail in Rett syndrome?
Some microglia may develop an aversion to housekeeping long before old age. Studies by Jonathan Kipnis of the University of Virginia School of Medicine in Charlottesville and colleagues have found a link between faulty microglia and Rett syndrome, a severe autism spectrum disorder that strikes in early childhood.
Rett syndrome affects motor skills and often leaves young victims wheelchair bound, unable to talk or use their hands and gasping for breath. The syndrome is caused by mutations in a single gene, called MECP2, on the X chromosome. Because they have only one X chromosome, boys born with the mutation usually die within weeks of birth. Girls with one faulty copy generally begin to show symptoms of decline before 18 months of age.
Microglia are suspects in causing some of the dysfunction because the brains of mice with Rett-like disease become cluttered with cellular debris, according to Kipnis. In a study published last year in Nature, he showed that replacing faulty microglia with healthy cells made a difference in mice with the disease.
Microglia are the only cell type in the brain that can be “feasibly” replaced, Kipnis says. That’s because microglia are part of the immune system, and bone marrow transplantation results in a partial replacement of these cells.
First, Kipnis’ team exposed 4-week-old mice with Rett-like symptoms to radiation to wipe out their existing immune cells, including microglia. Then the team injected the animals with bone marrow cells that had a working copy of the MECP2 gene.
The treatment reduced and slowed progression of the symptoms in both males and females. Males grew stronger, gained weight and began to walk. Females also showed improvement in walking and breathing.
To be sure that the improvements were because of microglia in the brain and not immune cells elsewhere in the body, the scientists gave bone marrow transplants to Rett mice that did not get a dose of radiation to their brains, sparing the faulty microglia. The transplant did nothing for these mice.
Though unconventional, the approach shows how treatments focused on the immune system can correct problems in the brain, Kipnis says.
“What drives our interest about the immune system, and microglia in particular, is that even though neurological disorders are disorders of neurons, it’s impossible to treat them directly,” he says.
“We can’t fix neurons, we can’t replace them and we can’t change them.”


{kind=link}