Nerve cell links severed in early stages of Alzheimer’s
Synapse loss study points to new path for fighting degenerative brain disease
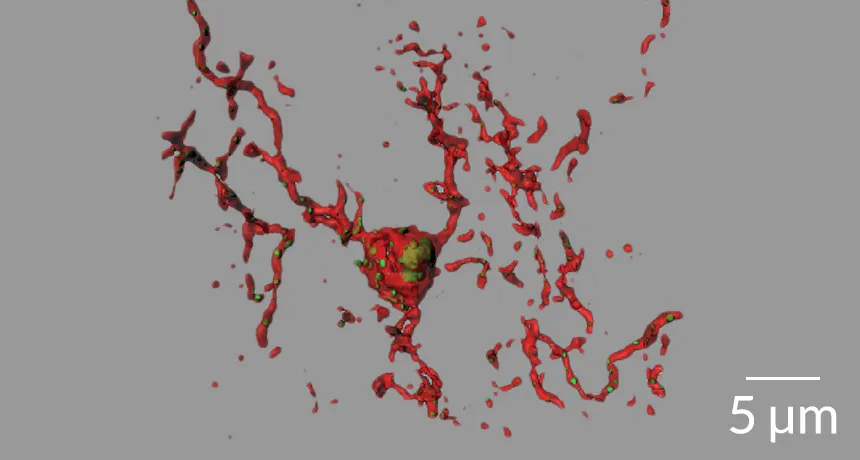
CRIME SCENE Following an injection of the Alzheimer’s-related protein amyloid-beta, a microglial cell (red) engulfs synapses (green) in a mouse’s hippocampus.
S. Hong et al/Science 2016
In the early stages of Alzheimer’s disease, an overzealous set of proteins and cells begins to chew away at the brain’s nerve cell connections, a study in mice suggests.
That finding, described March 31 in Science, adds to a growing body of research that implicates excessive synaptic pruning, a process that shapes the young brain by culling unused connections, with disorders later in life. The new work pins the loss of nerve cell–connecting synapses on particular immune system molecules and a notorious Alzheimer’s-linked protein.
By uniting these multiple strands of evidence, the study may help explain the earliest steps in Alzheimer’s march of neural destruction. “No one has put it together in quite this way,” says neuropathologist John Trojanowski. If the same process happens in humans, the new results may point to ways to slow or stop Alzheimer’s, says Trojanowski, of the University of Pennsylvania’s medical school.
A curious observation led to this new view of neural whittling. A protein called C1q was packed around synapses in the brains of young mice genetically engineered to show signs of Alzheimer’s. And C1q was most abundant in brain areas known to suffer synapse losses as Alzheimer’s takes hold.
C1q is a member of the complement cascade, a group of immune system proteins that calls in microglia cells to gobble up synapses or cells. This pruning is essential as the brain develops. But these neural gardeners seem to spring back into action in the early stages of Alzheimer’s, neuroscientist Beth Stevens of Boston Children’s Hospital and Harvard University and colleagues found. And that reactivation seems to be helped along by the Alzheimer’s-related protein amyloid-beta.
In the brains of mice that weren’t genetically engineered, injections of oligomeric A-beta, the form thought to be the most dangerous, caused C1q levels to rise. Along with this increase, synapses got destroyed, the team found. But A-beta injections didn’t harm synapses in mice lacking C1q, showing that C1q and A-beta are both needed for excessive pruning. How the two proteins exactly work together isn’t clear, Stevens says, but “they are definitely there at the right time and the right place.”
Complement proteins and microglia are known to be active in late-stage Alzheimer’s, when the inflamed brain is packed with sticky gobs of A-beta. But the new results suggest that the synapse-pruning pathway is active much earlier in the disease process, long before A-beta plaques form. “The story is extremely compelling and tight in Alzheimer’s mouse models,” says neurologist Scott Small of Columbia University.
There are reasons to think that a similar process happens in people. Autopsy studies by neurobiologist Stephen Scheff of the University of Kentucky in Lexington and colleagues, for instance, have turned up fewer synapses in the brains of people with mild cognitive impairment — thought to be an early stage of Alzheimer’s. The cause of that synapse loss could certainly be explained by changes in complement proteins or microglia, Scheff says.
Any therapy that would target this pruning process would first depend on identifying people at risk. And so far, there are no good tests to spot excessive oligomeric A-beta in the brain, says neurologist Sam Gandy of Mount Sinai Medical Center in New York City. “Oligomers are invisible,” he says.
But if screening methods are developed, then the prospect of stopping Alzheimer’s by stopping synapse loss is appealing, Small says. A drug that could prevent C1q or its conspirators from targeting synapses for destruction might halt the damage, for instance. “It’s easier to cure a sick cell than a dead cell,” he says.
Overactive synaptic pruning may be behind other brain disorders, Stevens suspects. She and her colleagues recently implicated a different complement cascade protein in schizophrenia (SN: 2/20/16, p. 7). “This may be a pathway that is dysregulated and playing a role in synapse loss in a host of neurological diseases, not just one,” she says. Stevens and several coauthors are involved with a company that is developing a drug to block C1q.