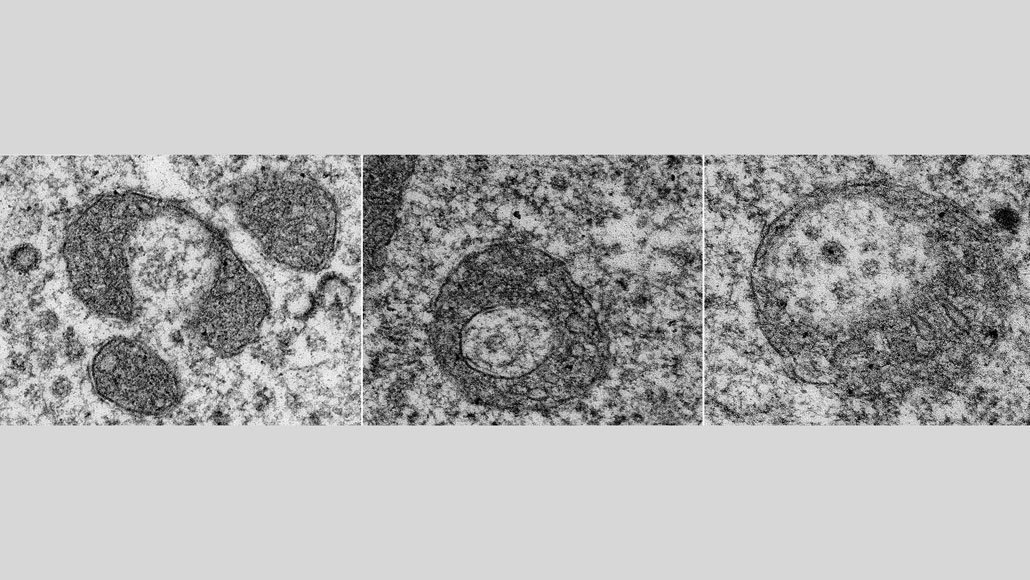
A newfound type of mitochondrial destruction starts when one of the cellular power plants stretches out and bends into a U (left). Its ends connect and fuse into a ring (center), and the organelle begins dismantling itself from the inside out (right).
Mukesh Gautam