Take a Breath: Fatty substance may play role in cystic fibrosis
Accumulation of a fatty compound called ceramide in the lungs could set the stage for the chronic lung infections of cystic fibrosis, a study in mice suggests. The finding offers a new twist on the still-unresolved course of this hereditary disease.

Patients with cystic fibrosis have a mutation in the CFTR gene. The protein normally encoded by this gene shuttles ions in and out of cells, ushering needed chemicals through the cell’s membrane. But because cystic fibrosis patients’ CFTR protein is missing or defective, the shuttling goes awry and sets off a process that leaves patients’ lungs cluttered with mucus. The ultimate result is the respiratory failure characteristic of this disease.
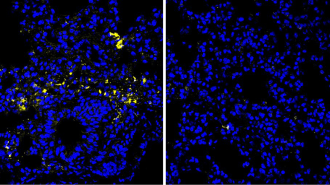
Earlier studies had shown that a faulty or missing CFTR protein could make cells more alkaline. In the new study, physician Erich Gulbins of the University of Duisburg-Essen in Germany and his colleagues observed a rising pH over time in the cells of mice genetically engineered to lack a functional CFTR gene. The higher pH resulted in an enzyme imbalance that caused cells to overproduce ceramide.
Excess ceramide accumulation caused inflammation and cell death in the animals’ lungs. When the scientists exposed the mice to a bacterium called Pseudomonas aeruginosa that commonly infects cystic fibrosis patients, mice lacking a functional CFTR protein proved significantly more susceptible to it than normal mice. But when the engineered mice received the drug amitriptyline, they fended off P. aeruginosa and survived longer than mice not getting the drug, the authors report in the April Nature Medicine.
Amitriptyline is an antidepressant that received regulatory approval in 1983. In these new tests it inhibited the enzyme that synthesizes ceramide, returning ceramide levels to near-normal. With ceramide under control, the mice were able to beat back P. aeruginosa.
The mucus in the lungs of cystic fibrosis patients provides cover for infective agents such as P. aeruginosa, says Gerald Pier, an immunologist at Harvard Medical School and Brigham and Women’s Hospital in Boston. His team has found that in healthy people, cells with normal CFTR bind to P. aeruginosa and dispatch the microbe routinely. But in cystic fibrosis patients, the bacterium dodges this binding and evades immune detection. The microbe lingers in the mucus and causes long-term infection, he says.
The German team tested cells obtained from cystic fibrosis patients’ lungs and nasal cavities and found they contained four times as much ceramide as did similar cells taken from healthy individuals.
Ceramide “could play a significant role,” says study coauthor Anna van Heeckeren, a veterinarian at Case Western Reserve University in Cleveland. “These are intriguing data and fascinating results, but I don’t think it’s the ultimate answer” to the cystic fibrosis puzzle, she says.
Pier agrees that the biological course of cystic fibrosis is far from clear. But the findings might lead to a clinical trial testing amitriptyline in cystic fibrosis patients, despite the drug’s history of side effects that include dizziness, headache, dry mouth, and diarrhea.