You, in a Dish
Cultured human cells could put lab animals out of work for chemical and drug testing
At 8 o’clock on a March morning last year, doctors at Northwick Park Hospital in London began injecting six healthy men with an experimental arthritis drug. It was the drug’s first safety trial in humans, and it had passed all the necessary tests on mice and monkeys with no indication of danger.

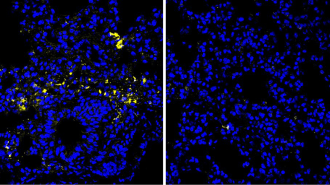
Once inside each man’s bloodstream, the drug bound strongly to the “seek and destroy” cells of the immune system, the T cells. As a result, these attack cells became hyperactive and began leaving the blood vessels and entering tissues—something T cells normally do only at the site of an infection. The T cells proceeded to attack and kill healthy cells of vital organs such as the heart, liver, kidneys, and lungs.
Within an hour, the men were vomiting and writhing in excruciating pain as their immune systems began attacking their bodies from the inside out. All six nearly died, and they still suffer from lingering health impairments.
Clearly, the animal tests had missed something.
As invaluable as mice have been for medical research, differences do exist between human and mouse biology. Sometimes, these differences generate misleading test results. In the case above, for example, the drug bound to a receptor molecule on the human T cells more tightly than it did to the mouse version of that receptor. The drug also activated a kind of T cell that the lab mice lacked.
A better way to find out how a drug will affect human cells, some scientists say, is simply to test it on human cells. That’s now becoming a practical option.
Over the past few years, scientists have developed sophisticated ways to screen drugs and other compounds on lab-grown cells from various human organs. Coupled with the burgeoning knowledge of cells’ inner workings that comes from genomics, proteomics, and other “-omics,” these screening techniques offer a way to test compounds on human biology long before they’re tested on actual humans.
These screens could aid pharmaceutical development by identifying promising candidates and weeding out compounds earlier if they don’t work in humans (even though they may work in mice). Many scientists think that in the wake of genomics, such screens are the next logical step.
“Everybody in the industry thinks this is the right way to go,” says Aled M. Edwards of the Ontario Cancer Institute in Canada, who has no connections with companies offering such screening services. “I know that pharmaceutical companies are doing this.”
And drug companies aren’t the only ones. The Environmental Protection Agency and the National Institutes of Health are launching a joint program to test potentially toxic industrial chemicals on human cell cultures instead of on animals, the agencies announced in February during a meeting in Boston of the American Association for the Advancement of Science, as well as in the Feb. 15 Science (SN: 2/23/08, p. 117).
Sparing animals from serving as guinea pigs and improving the accuracy of test results are both reasons for the growing interest in these screening techniques, but the pharmaceutical industry has another reason as well. Money.
Playing the numbers
The cost of developing new drugs has been steadily increasing for decades, yet the number of new drugs that make it to market each year has remained essentially constant. As a result, the average development cost for each new, approved drug exceeds $800 million, by some estimates.
“Plenty of drugs are going into the pipeline—it’s just that too many are dropping out,” says Janice M. Reichert of the Tufts Center for the Study of Drug Development in Boston. Many drugs make it through the gauntlet of laboratory and animal tests successfully, only to fail during human trials. Some drugs simply prove to be ineffective in people; others turn out to be unsafe or to have unacceptable side effects.
Recruiting people to serve as subjects and running clinical trials can cost tens of millions of dollars. So when a drug fails near the end of trials, the financial loss can be enormous.
In a sense, this rising dropout rate is a consequence of the genetics revolution. In the 1970s, pharmaceutical companies adopted shotgun tactics for screening thousands of chemicals using high-throughput chemistry. The search focused on compounds that hit the same protein targets as did existing, proven drugs. Researchers tested any matching compounds on animals in the hope that the drug was more effective and had fewer side effects than the old drug, often with great success.
The rise of human genetics yielded a windfall of novel biological targets for treating diseases, and companies applied the same shotgun techniques to find drugs for these new targets. The effects of these drugs, however, were less predictable because no previous drugs bound to the same targets. Scientists relied on animal tests to show that a drug worked and to reveal dangerous side effects, but differences between animal and human biology meant that some effects of the new drugs were inevitably missed.
“What’s going in the pipeline is perhaps less validated than in the past,” Reichert says. “Not as much is known about them.”
Tests on human cell cultures could help to fill that knowledge gap, many scientists believe.
The concept is straightforward: Grow cells from various human tissues in the lab, add a dose of the compound, and measure the resulting changes in the cells’ activities.
Dosing and measuring are performed en masse by automated tabletop machines. These use standard laboratory techniques to monitor what happens to individual functions, such as insulin production, or to record changes in the amounts of proteins known to be good indicators of the cells’ inner workings. Computers can immediately sift the resulting data and pop out a response profile for the compound.
Two innovations in recent years have made this approach possible. The first is the explosion of knowledge that resulted from the sequencing of the human genome. Trying to make sense of this torrent of data has spawned the field of systems biology, an attempt to put all the genetic pieces together and understand how the cell operates as a whole. Systems biologists produce complex maps of how genes and proteins interact, and these maps help scientists to analyze results from a drug screening.
The other advance is less sexy but perhaps more important—learning how to grow human cells in the lab so that they continue behaving like human cells.
“When you take the cells and put them in the dish, you lose all their specialized functions,” says Mina J. Bissell of Lawrence Berkeley National Laboratory in Berkeley, Calif. That’s because cells “talk” to each other by passing chemical signals back and forth. They also sense their physical surroundings through proteins on their surfaces called integrins. All these cues serve to orient the cells in the body and inform them about how to behave so that they cooperate with the rest of the cells in the tissue.
“The cells are not complete by themselves. They need signals from outside,” Bissell says. “The unit of function literally is the tissue.”
Pharmaceutical companies have tested compounds on human cells for decades, of course, but these tests have involved only simple cultures of one type of cell. Typically, the goal has been merely to check for blatant toxicity. Researchers expose the cells to a drug candidate and observe whether the cells continue growing or die.
Such tests are far simpler than these new screening techniques, which attempt to extract much more information from the cultured cells than just a live-or-die response. To do so, scientists must grow cells in conditions that mimic life inside the body. Often, that entails growing several different types of cells from a tissue in a single dish so that the cell types can “socialize.” Researchers also add hundreds of special chemical factors that are normally present in the tissue milieu, such as signaling molecules called cytokines.
“It’s been a slow process to refine techniques for growing cells so they retain their normal behavior,” explains Ellen L. Berg, chief science officer and cofounder of BioSeek, a biotech startup in Burlingame, Calif., that tests compounds on cultured human cells. “There was really no one breakthrough. It has just taken a long time to learn how to grow human cells.”
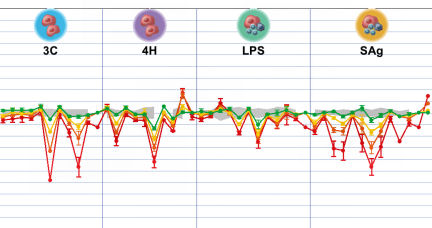
To confirm that cells in these more advanced cultures truly feel right at home, Berg and her colleagues checked whether the cultures responded to older drugs in the same ways that people’s tissues do in the clinic, and whether those responses were repeatable.
Berg’s team combined endothelial cells and white blood cells called leukocytes, and then added a carefully prepared recipe of signaling molecules. The researchers placed small samples, each containing about 1,000 to 10,000 cells, into tiny wells in laboratory plates. Each plate is only a few inches across and contains several hundred wells arranged in a grid. Automated machines then exposed these samples to a long list of well-studied drugs, including old standbys such as the anti-inflammatory ibuprofen. The resulting changes in key protein concentrations closely matched the responses of cells in people, the team reported in the January-February 2006 Journal of Pharmacological and Toxicological Methods. Repeating the screens produced similar results.
Enter the matrix
For rapid, high-throughput techniques, time constraints often limit scientists to making flat, 2-D cell cultures such as those used by Berg’s team. But some scientists are taking mimicry of real tissues a step further by growing cells in 3-D matrices.
Embedding cells in a medium that contains nutrients as well as a mesh of fibers such as collagen simulates the environment that surrounds cells when they’re within a tissue. For some diseases, cues from that 3-D environment make a big difference.
For example, breast cancer cells grown in 3-D cultures develop resistance to chemotherapy drugs at a similar pace as they do in patients, Valerie M. Weaver of the University of California, San Francisco reported in Washington, D.C., at the December 2007 meeting of the American Society for Cell Biology (SN: 12/15/07, p. 382). Cancerous cells grown in conventional cultures become resistant to drugs more slowly, giving doctors a false sense of security, Weaver says.
But the 3-D environment isn’t always essential, says Bissell, a pioneer of 3-D cell culture techniques. “What we find is that some drugs more or less behave similarly” in 2-D and 3-D cultures, “while some drugs behave very differently,” she says.
Even with the most realistic cultures, tests on cells outside the body will always have some limitations, of course. “Nothing that’s done pre-clinically will assure with 100 percent certainty what you’ll see in a human being,” Reichert says. “There are just too many unknowns.”
And regardless of which kind of culture scientists use, they must somehow decide which indicators will best tell them how the cells are responding to the test compound. To choose which proteins to monitor—and to interpret the results—scientists draw from traditional clinical data on patients with the disease and from the relatively new discipline of systems biology.
If the Human Genome Project was a blockbuster movie, systems biology is the sequel.
Revenge of the genome
Once scientists had the human genome’s enormous parts list of roughly 25,000 genes in hand, next came the daunting task of assembling all those pieces and trying to understand how they work together as a whole.
After all, it’s the net effect of all those interacting parts that produces the behaviors of cells, organs, and people. These byzantine interactions vary in sickness and in health, from tissue to tissue, from cell to cell, and over time. Understanding the dynamics of all these interactions in all these scenarios is the goal of systems biology.
“It came out of the problems that followed from doing the human genome. Because once you have all this information, what are you going to do with it?” Berg says. “The human genome gave us more questions than answers.”
To make sense of the genome, systems biologists think in terms of networks. If two kinds of proteins or other biological molecules interact, they are connected on the network. Link by link, these connections reveal the web of interacting proteins that form metabolic pathways. Each protein in a pathway plays a role in multistep cell processes such as breaking down glucose or making insulin.
These network diagrams also show how individual pathways crisscross to form a tangled web. Each protein in a pathway can interact with molecules in other pathways, sometimes dozens of them. “There is no such thing as a single pathway,” says Eugene C. Butcher, a pathologist at Stanford University and cofounder of BioSeek. “There’s only a complex network, and this network topology changes in different conditions.”
By mapping this web of connections, scientists can see that a drug designed to target a protein in one pathway could also have unintended consequences for other pathways. Also, if a drug produces a bewildering pattern of increased and decreased protein activity, scientists can look at the links among the proteins and identify key places on the map where the drug must be acting to produce the observed effects.
Such techniques run counter to established ways of thinking at large pharmaceutical companies, which have traditionally been focused on the biochemistry of individual drug targets. “If you look at any pharma company, biologists are low on the totem pole,” Berg says. “Chemists rule the roost.”
But even Big Pharma has started to dabble in these new screening techniques. Eli Lilly and Co., based in Indianapolis, has started a systems biology-based research department that tests compounds on human cells. And Butcher says that his company routinely screens compounds for three of the five largest pharmaceutical companies, which he declined to name.
“Drug companies definitely want to try it,” says Edwards of the Ontario Cancer Institute, “because human cells are certainly better than rat cells.”