Boning Up
Turning on cells that build bone and turning off ones that destroy it
Construction and demolition crews working side by side. It sounds insane, but that’s how the human skeleton grows and maintains itself. Each of its 206 bones harbors cells that continually deposit a rigid protein matrix that becomes mineralized. Each bone also contains cells that can break that structure down.
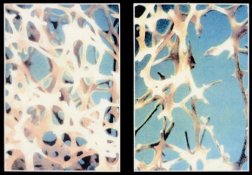
The bone builders keep ahead of the bone destroyers as a child grows, but the balance can shift as a person gets older.
Bones weaken when osteoclasts, the matrix-resorbing cells, outpace osteoblasts, the matrix-making cells. A wide variety of conditions—most notably osteoporosis, but also rheumatoid arthritis, leukemia, and HIV infection—shift the cellular tug of war in favor of the bone destroyers. The weak bones that result can suffer debilitating, even deadly, fractures.
Scientific advances on two complementary fronts now offer new hope in the body’s battle against bone loss. In one of the most exciting stories in bone research in many years, scientists have unearthed a fundamental mechanism by which oseteoblasts communicate with osteoclasts. Medical researchers are already testing a new drug that exploits this knowledge to stop bone deterioration.
In a more surprising development, popular cholesterol-lowering drugs known as statins seem to stimulate growth of new bone. Statins have thickened the bones of mice in laboratory experiments, and test-tube studies suggest a mechanism for this action. However, it’s not clear yet whether statin drugs will jump-start bone growth in people.
Unusual skeletons
The excitement began in 1996 when X rays of some genetically engineered mice revealed something unusual about their skeletons. “We found that [the animals] had extraordinarily dense bones,” recalls Colin R. Dunstan of Amgen, a biotech firm in Thousand Oaks, Calif.
He and his colleagues at the company had been puzzling over the role of a protein encoded by a gene that they had recently discovered. As part of their investigation into the protein’s function, the investigators created mice with extra copies of the gene. The animals synthesized abnormally large amounts of the protein, which somehow led to the thicker-than-normal bones.
The puzzling protein—dubbed osteoprotegerin (OPG) by Amgen because it seemed to protect the integrity of bones—turned out to be a key part of the molecular conversation between osteoblasts and osteoclasts. While the goals of these two cell types appear at odds, scientists have long known that the cells cooperate.
“The cells that make the bone also control its removal or resorption,” says Dunstan. How the two types of cells communicate had been a mystery until studies in the past few years revealed that OPG binds to a protein called OPG-ligand. This attachment prevents OPG-ligand from rousing osteoclasts into action.
“OPG-ligand is like the accelerator of your car. If you step on OPG-ligand, you lose bone. OPG is the brake of the system. If you step on OPG, then you have more bone. The balance between the two determines how much bone we have,” says Josef M. Penninger of the University of Toronto.
Osteoblasts make both OPG and OPG-ligand, scientists have learned. OPG-ligand binds to surface proteins on osteoclasts, triggering proliferation of these cells and increasing their bone degradation. OPG secreted in the bone matrix serves as a decoy, sequestering OPG-ligand and preventing it from triggering bone resorption.
Since many proteins in the body stimulate osteoclast activity, investigators wanted to confirm the importance of OPG-ligand by creating mice lacking its gene. In the Jan. 28, 1999 Nature, Amgen scientists and Penninger’s team reported that like the mice producing excess OPG, mice without OPG-ligand develop unusually thick bones. The latter mice have no osteoclasts at all.
“OPG-ligand is the essential molecule for osteoclast development,” says Penninger. By controlling the balance between OPG and OPG-ligand, he speculates, “you can treat hundreds of diseases that have associated bone loss.”
These molecules do seem to make up “a key and previously unrecognized system in the regulation of osteoclast genesis,” agrees William J. Sharrock of the National Institute of Arthritis and Musculoskeletal and Skin Diseases in Bethesda, Md.
Natural body chemicals
A variety of natural body chemicals and drugs leads to bone loss when administered to animals or people. Scientists are now finding that almost all of those substances slash production of OPG, boost creation of OPG-ligand, or do both.
Consider the glucocorticoids, a class of steroids that includes prednisone. Prescribed for a variety of medical reasons, these drugs sometimes set off quick and dramatic bone loss.
Treating laboratory-grown osteoblasts with such steroids inhibits their synthesis of OPG and heightens their OPG-ligand production, Dunstan and other researchers from Amgen and the Mayo Clinic in Rochester, Minn., reported in the October 1999 Endocrinology.
In the September 1999 Endocrinology, some of the same scientists showed that applying estrogen to human osteoblasts stimulates activity of the gene for OPG. This may explain why an aging female’s diminishing estrogen production leads to bone loss and may account for the bone-protecting effects of hormone-replacement therapy. “Now, we have the molecular explanation for postmenopausal osteoporosis,” says Penninger.
The interplay of OPG and OPG-ligand may resolve another medical mystery. Rheumatoid arthritis, diabetes, multiple sclerosis, lupus, hepatitis, lymphomas, AIDS, and many seemingly unrelated diseases can produce osteoporosis along with their other symptoms. “Kids with leukemia are growth retarded because of bone loss,” adds Penninger.
The only thing the diseases seem to have in common is that they involve a class of immune cells called activated T cells. How do T cells, which normally fight infections, interact with bone? The first glimpse of an explanation came in 1997 from the Seattle biotech firm Immunex.
Scientists there described a protein made by activated T cells. That protein turned out to be OPG-ligand. The unanticipated connection between OPG-ligand and T cells grew stronger in 1998 when Penninger found that his mice lacking the gene for OPG-ligand had no lymph nodes, one of the tissues where T cells mature.
Scientists have since found that almost anything that stirs quiescent T cells into action also triggers their synthesis of OPG-ligand. Although no one can explain why the immune cells react this way, this link suggests that chronic infections, autoimmune disorders, and T cell cancers can all make osteoclasts go wild.
“Every time you turn on T cells, they make OPG-ligand, and this leads to bone loss,” says Penninger. “This explains hundreds of clinical studies.”
To buttress that argument, he and his colleagues recently tested whether OPG could slow the progression of an artificially induced rodent condition similar to rheumatoid arthritis. In patients with this autoimmune disorder, joint inflammation is followed by the destruction of surrounding bone and cartilage. Rheumatoid arthritis afflicts more than a million people in the United States, crippling many of them.
When signs of inflammation appeared in mice treated to develop arthritis, the investigators started giving the animals daily injections of OPG. The inflammation persisted, but the shots prevented bone and cartilage destruction, Penninger and his colleagues report in the Nov. 18, 1999 Nature.
They were pleasantly surprised that OPG blocked cartilage as well as bone damage, particularly since scientists aren’t sure how rheumatoid arthritis destroys the cartilage in people. “The simplest explanation is the cartilage breakdown has been a matter of the underlying bone deteriorating,” notes Sharrock. An alternative explanation is that OPG-ligand may stimulate osteoclasts to degrade cartilage as well as bone, says Sharrock, or it may incite other cells to destroy the cartilage.
The long trek
Moving biological advances from the laboratory bench to the doctor’s office can take years and frequently fails, but Amgen has already begun the long trek with OPG. The company’s first step was to modify the protein so that it circulates in the blood much longer than the natural version does.
Last September, at the American Society for Bone and Mineral Research meeting in St. Louis, Dunstan and his colleagues announced results of initial safety tests. Forty women who had just gone through menopause received a single shot, in varying doses, of the modified OPG. Physicians documented no side effects other than some minor irritation at the injection site.
While the researchers didn’t design this initial trial to test the drug’s efficacy, they did monitor daily the rate of bone resorption in the women by measuring a bone-breakdown product in their urine. At the highest dose, the single shot of modified OPG reduced a woman’s normal bone resorption rate by as much as 85 percent. The drug’s effect lasted for up to a month, says Dunstan.
Amgen now plans larger trials of the drug with postmenopausal women, as well as with people suffering localized bone destruction due to metastatic bone cancer. As for rheumatoid arthritis, specialists on the topic warn that the rat condition studied by Penninger and his colleagues doesn’t perfectly model the human disease.
“It has a lot of shortcomings,” cautions Gary S. Firestein of the University of California, San Diego. He adds that other T cell-based strategies have failed to help people with rheumatoid arthritis. Still, he believes that testing OPG on people with the autoimmune disorder is warranted. Indeed, Amgen may begin such a trial this year.
Penninger admits that he can barely contain his enthusiasm about using OPG to stop bone loss in osteoporosis and T cell disorders. He compares its potential significance to the realization that giving insulin to diabetics prevents the most serious consequences of the disease. “I think it’s the insulin for the bone,” says Penninger. “I’ve never been as confident in my whole scientific life.”
Growing new bone
Stopping bone loss is one thing. Tricking the body into growing new bone is quite another. Physicians desperately need such a technique because they often don’t diagnose osteoporosis until bones have already become irrevocably weakened.
“The Holy Grail of osteoporosis research is a drug that will restore or rebuild bone that is lost,” notes Gregory Mundy, a researcher at the University of Texas Health Science Center and founder of the biotech firm OsteoScreen, both in San Antonio.
“We need something for the little old lady who has already lost 40 percent [of her bone mass],” agrees C. Conrad Johnston Jr. of Indiana University School of Medicine in Indianapolis, who is president of the National Osteoporosis Foundation.
The widely used statins may provide the answer to Johnston’s request. In the Dec. 3, 1999 Science, Mundy and his colleagues report that statins stimulate bone formation in test-tube studies and in experiments with rodents. Since physicians have prescribed statins for more than a decade and the drugs have a well-documented safety record, the unexpected results tantalize bone researchers.
The initial hint that statins have bone-building properties came when the scientists began looking for small molecules that trigger the maturation of osteoblasts. The researchers used automated machines to screen more than 30,000 natural compounds for their ability to activate a gene encoding a molecule—bone morphogenetic protein-2, or BMP-2—that promotes local bone formation.
Mundy’s team found just one molecule, lovastatin, that turned on the gene for BMP-2. Startled by this unanticipated result, the researchers quickly tested whether other statins, not among the compounds they originally screened, would also stimulate BMP-2 production. All but one did so.
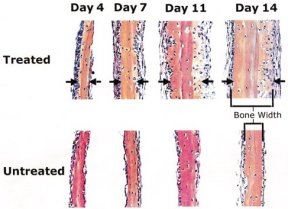
The investigators next examined whether statins can influence bone creation. By adding the drugs to laboratory dishes in which pieces of mouse skulls were growing, they showed that statins could double or triple the amount of new bone formation. The statins seem to stimulate activity, proliferation, and maturation of osteoblasts.
Mundy and his colleagues then began testing statins on animals, initially injecting the drugs three times a day into tissue above the upper part of the skull of mice just a few weeks old. Five days of such injections produced 50 percent more bone than normal.
Since part of the appeal of the statins is that people can take them as pills rather than as injections, the scientists also administered oral doses of the drugs to female rats. In rodents that have had their ovaries removed, osteoporosis normally occurs quickly. When the researchers examined such rats after they had taken statins for a month, those on the highest doses had double the bone-formation rate of untreated rats, Mundy and his colleagues report.
Hans Oxlund of the University of Aarhus in Denmark and his colleagues have confirmed the bone-building properties of statins in rodents. Compared with adult female rats receiving a saline solution, those getting statin had twice as much bone formation over 3 months, Oxlund reported at the St. Louis meeting of the American Society for Bone and Mineral Research. This extra bone has normal properties, Oxlund told Science News.
Statins slash cholesterol production in the body by inhibiting an enzyme that creates a cholesterol precursor. Laboratory results suggest that the drugs also increase bone production by inhibiting that same enzyme. Pharmaceutical firms may ultimately need to develop statin-related drugs that increase bone formation while allowing the body to continue making the cholesterol that all animal cells need, Mundy notes.
Yet companies probably won’t go to such lengths until they’re certain that statins build bone in people. Since human and rodent bone metabolisms differ significantly, the jury is still out on that question.
In an attempt to address the issue, Mundy, along with Steven R. Cummings of the University of California, San Francisco, recently analyzed bone density and hip-fracture data from about 600 women over the age of 55 who had participated in trials of statins’ effects on cholesterol. They were compared with similarly aged women taking other cholesterol-lowering agents.
The results were not conclusive, says Mundy, who calls upon the makers of statins to launch clinical trials specifically designed to test the bone-forming abilities of their drugs. Even such trials may not resolve the issue. Drug developers selected the current statins for their ability to move quickly from the bloodstream to the liver, the primary site of cholesterol synthesis, rather than to the skeleton. Statins that target bone may be needed, but such drugs could lose the benefit of the current statins’ enviable record. “You might have to start all over again on safety and toxicity,” says Sharrock.
While physicians don’t yet have mastery over the skeleton’s cellular construction and demolition crews, bone researchers are finally providing them with the commands they need to instruct their skilled workers.