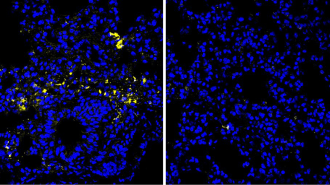
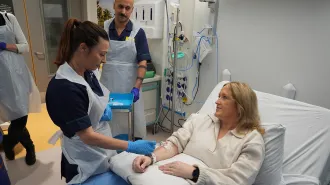
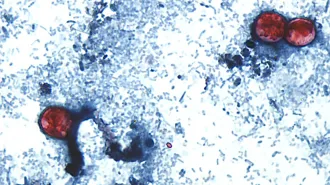
Treating cystic fibrosis patients before birth could safeguard organs
A study in ferrets hints an early start with a drug therapy may protect pancreases and lungs
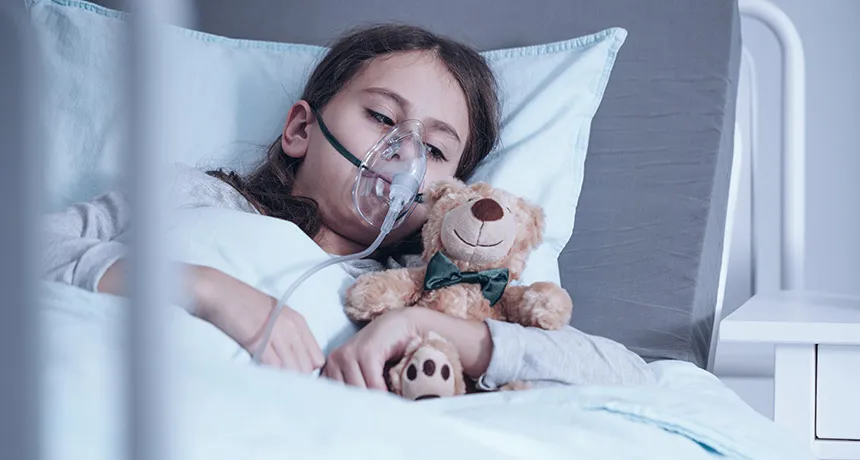
WHOLE BODY HELP Children with cystic fibrosis can have lung disease, failed pancreases and fertility problems. Research in ferrets suggests that treating patients with a drug in the womb may protect against those issues.
Photographee.eu/Shutterstock