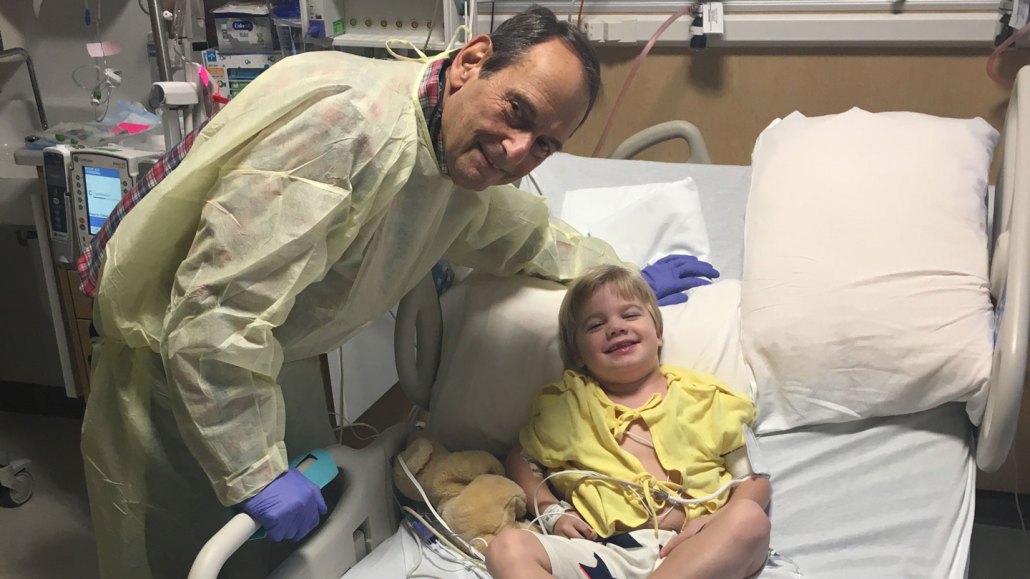
In 2018, Will Ungerer, shown with doctor Jerry Mendell at Nationwide Children’s Hospital in Columbus, Ohio, received experimental gene therapy for Duchenne muscular dystrophy, a disease that breaks down muscle.
S. UNGERER
Will Ungerer isn’t a typical fourth-grader.
The 10-year-old from Midlothian, Va., plays tag with his friends and swims in the ocean. “I think his peers would describe him as someone who is kind, sweet and funny, and supersmart,” says his mother, Sheila Ungerer. He won a countywide citizenship award and was named classroom student of the year.
But “he’s not a really fast runner,” his peers might say. “He doesn’t really run.” Will was born with Duchenne muscular dystrophy, a genetic disease that causes muscles throughout the body to break down over time. That includes not only skeletal muscles, but also the heart and diaphragm, which controls breathing. Eventually those important muscles stop working properly, leading to death. The disease is caused by mutations in the gene responsible for dystrophin, a protein that acts as a shock absorber to prevent damage as muscles contract. It affects an estimated 4.8 out of 100,000 people worldwide, mainly boys and young men. About 10,000 people in the United States have Duchenne muscular dystrophy.
Will celebrated his birthday last year at a laser tag park, his mother says, but other kids his age with Duchenne’s “don’t ride the bus. They don’t carry their tray in the lunchroom.”
The difference is that Will received an experimental gene therapy when he was 5 years old. Just before Christmas in 2018, doctors at Nationwide Children’s Hospital in Columbus, Ohio, gave Will an infusion of viruses. Those viruses delivered instructions to his muscles for making a short form of the dystrophin protein. Scientists at Sarepta Therapeutics in Cambridge, Mass., the company that makes the gene therapy, developed this version, called microdystrophin, to act as a replacement, hopefully protecting muscles from harm.
Though the therapy did not show statistically meaningful improvements compared with a placebo in one randomized controlled trial and results of an ongoing clinical trial to determine efficacy aren’t yet published, Will and the 83 other boys in Sarepta’s clinical trials now make the microdystrophin protein. Based in part on that evidence, the U.S. Food and Drug Administration approved the therapy earlier this year for use in 4- and 5-year-olds, and the first child to get the therapy after approval was infused on August 2.

For Sheila Ungerer, there’s no question that the gene therapy is working for Will. He now does everyday things he couldn’t before, such as climb stairs easily and brush his teeth and get dressed on his own. He can ride a bike and swims up to 500 meters at swim team practice.
A few months after the infusion, Sheila overheard Will talking to his younger brother Adam. “I was just outside the door, and he said, ‘Adam, remember when my legs used to hurt all the time? They don’t hurt anymore,’ ” she recalls. “That was his experience of life with inflamed muscles in his legs that were being damaged constantly. And then for that to subside is just an immeasurable impact on his life.”
The FDA has approved seven other gene therapies for rare genetic diseases, all since 2017. Each puts a healthy copy of a gene into cells to compensate for a missing or mutated one. Some potential treatments for rare diseases that use gene editing, a type of gene therapy that makes targeted changes at the DNA level, may soon win approval too.
Around the world, more than 2,000 gene therapies are in development, according to the American Society of Gene and Cell Therapy. That figure includes cells that have been genetically engineered to fight diseases, such as immune cells called CAR-T cells programmed to kill cancer or keep lupus from attacking the body (SN: 2/2/22; SN: 9/15/22). But unlike these therapies for cancer and autoimmune diseases — which could treat potentially millions of people, making them financially attractive to drug companies — each gene therapy for a rare disease may help thousands of patients or fewer.
Since gene therapy was first proposed to treat such genetic diseases in the 1970s, it has had thrilling highs — including the first successful gene therapy, in 1990, in a 4-year-old born with severe combined immunodeficiency, or SCID — and deeply troubling lows. Clinical trial participants have ended up with cancer and other serious health complications and have even died. The biggest problem appears to be the viruses used to ferry replacement genes where they need to go. Viruses are the obvious choice for delivery: They can carry the replacement gene in their own DNA or RNA and have built-in mechanisms for getting into cells. But viruses can’t always get the genes to the right cells, can slip their cargo into the wrong spot in the DNA and can trigger the immune system to set off deadly inflammation.
Challenges and setbacks, including the death of a teenager named Jesse Gelsinger in 1999, nearly derailed gene therapy. But researchers, companies and patient advocacy groups persevered. Today, researchers are finding new ways to tackle their delivery dilemmas. Some are developing better viruses or nonviral ferries, while others are using new tools to repair or replace damaged genes in place. One technique, tested so far only in mice, relies on technology similar to what has been used in COVID-19 vaccines.
Some researchers say the outlook has never been brighter for gene therapy for a wide variety of inherited diseases. Others are frustrated by the slow progress.
What’s the current state of gene therapy?
When scientists discovered the gene that is mutated in cystic fibrosis in 1989, gene therapy was just about to be demonstrated in clinical trials in people. “We were all saying, ‘There’s going to be gene therapy next year,’ ” says Garry Cutting, a geneticist at Johns Hopkins University who studies the disease.
But that prediction didn’t pan out. There have been at least 36 gene therapy trials involving more than 600 people with cystic fibrosis, none with lasting success, researchers reported in 2022 in Frontiers in Pharmacology.
“And what’s the problem? The problem has always been delivery,” Cutting says. “How do you get it in the right place?”
For cystic fibrosis, which affects more than 160,000 people worldwide, the right place is the lungs, where mutations in a gene called CFTR result in the buildup of sticky mucus that makes it hard to breathe. Numerous clinical trials have attempted to slip a healthy copy of the gene into stem cells that replenish the airway lining. But it’s not easy for viruses to get through the sticky mucus, past patrolling immune cells, through layers of unrelated cells and into the stem cells. “It’s a harsh environment. It’s a tough problem,” Cutting says.
Because of similar struggles, many of the first gene therapies to win approval instead harvest a patient’s blood-forming stem cells, grow and genetically modify the cells in the lab and then return the modified cells to the patient, usually in the form of a bone marrow transplant. That is also the approach that CAR-T cell therapies for cancer use.
Editing cells outside the body, or ex vivo, has worked for rare diseases that affect the blood or immune system. Yet it comes with limitations, since scientists haven’t worked out ways to extract stem cells from tissues other than blood and bone marrow and replace them later. It also comes with its own dangers. It typically requires chemotherapy to kill existing bone marrow so the modified cells can take hold and replicate. And the viruses ferrying the replacement genes into the cells in the lab can still be problematic.
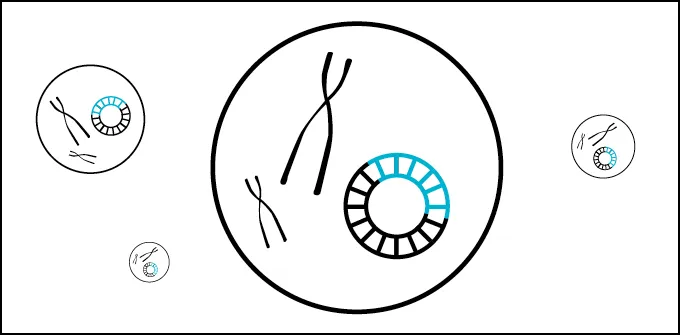
Ex vivo versus in vivo therapy
Many early gene therapies remove stem cells (circles) from a patient’s body, modify them with the help of viruses (hexagons) and then return the modified cells. This ex vivo approach often requires chemotherapy and only works for some diseases. In vivo therapies modify cells in the body, but getting the therapies to the right cells can be a problem.

Early gene therapies used retroviruses, which often insert their DNA cargo near active genes. Some children who got gene therapy in 1999 and 2000 for inherited diseases that disable the immune system developed leukemia when the inserted gene ramped up activity of nearby cancer-causing genes (SN: 1/14/03; SN: 7/11/13).
Most gene therapies today instead rely on viruses engineered to deliver their cargo to safer locations and to cause less of an immune response. But still, the track record is far from spotless.
In September 2022, for example, the gene therapy company bluebird bio, based in Somerville, Mass., received FDA approval for an ex vivo lentivirus-delivered gene therapy for children with a rare, fatal neurodegenerative disease known as cerebral adrenoleukodystrophy. About 1 in 4,000 to about 1 in 50,000 babies born will have adrenoleukodystrophy, and 35 percent to 40 percent of boys with the disease develop the severe form, which typically leads to death within five to 10 years if treatment doesn’t work. The approval came despite three cases of a type of cancer called myelodysplastic syndrome among clinical trial participants. The company determined that the lentivirus was indeed at fault and the therapy, called Skysona, comes with a warning about the cancer risk and recommendation for yearly cancer screening for 15 years.
Researchers have had successes with gene therapies that work in the body for some forms of blindness (SN: 5/24/21) and with a degenerative nervous system disorder called spinal muscular atrophy. Will Ungerer’s gene therapy also relies on an in vivo approach. In all three cases, the replacement genes are delivered by adeno-associated viruses, or AAVs. These viruses usually drop their cargo in the nucleus where it hangs out separately from the rest of a person’s DNA.
But the payload size of AAVs is limited, they may not infect all cells equally, and they can trigger dangerous and deadly immune responses. Patients who have gotten gene therapies delivered by AAVs have also developed liver damage or failure and microscopic blood clots that can lead to organ damage.
On top of all those risks, once the immune system of a patient receiving in vivo gene therapy makes antibodies and defenses against a delivery virus, redosing becomes difficult or impossible.
On March 22, 2018, Conner Curran of Ridgefield, Conn., then 7 years old, became the first person to get an experimental in vivo gene therapy developed by Pfizer for Duchenne muscular dystrophy. “Before gene therapy, he really struggled to go up the stairs,” says Conner’s father, Chris Curran. “Two months after gene therapy, he’s running up the stairs.”
Chris and his wife, Jessica, founders of the nonprofit Kindness Over Muscular Dystrophy, an organization that raises money for muscular dystrophy research, marveled at the improvements in Conner’s strength and agility. That’s just not something that happens for children with Duchenne muscular dystrophy, Chris says. “It’s a progressive muscle-wasting disease.… You don’t ever get better. You just get worse and worse and worse, and we actually saw him getting better.”
Though the improvement continued for three to four years, Chris and Jessica have observed a slow decline over the last year. “We knew going into this that it wasn’t necessarily a cure,” Chris says. He is hopeful that the gene therapy may still buy Conner and other boys time by strengthening their heart and diaphragm muscles, but Conner may need another dose of gene therapy to keep his skeletal muscles from deteriorating.
Unfortunately, now age 13, Conner has high levels of antibodies against the AAVs that delivered Pfizer’s version of microdystrophin to his muscles. He will be one of six children who previously got gene therapy to enter a clinical trial aimed at lowering their antibody levels against the AAVs.
Getting healthy cells in
All gene therapies for rare diseases that are approved today put a healthy copy of a gene into cells, but the therapies differ in how they get the gene in.
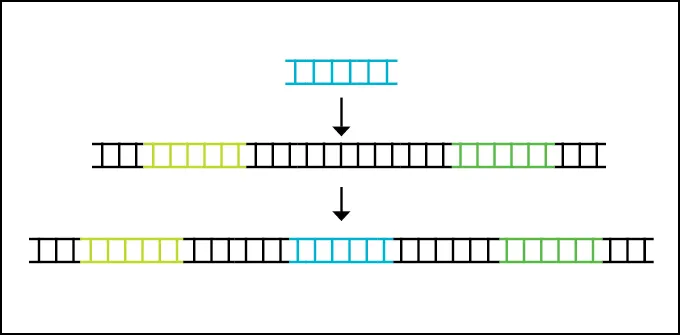
CRISPR gene therapy comes along
Even when a virus gets its cargo to the right cells — the right zip code — that doesn’t mean it’ll find the right address. Even today’s viruses don’t usually deliver the gene to its normal location in the DNA, says Matthew Porteus, a physician-scientist at Stanford School of Medicine. The delivery locations aren’t completely haphazard, but they’re not entirely predictable either. It’s a bit like ordering a pair of pants for store pickup at Old Navy. The pants will get to an Old Navy, but until they arrive, you won’t know exactly which one.
For some genes, it’s fine to place them elsewhere in the genome or let them hang out outside of chromosomes, Porteus says. Other genes have to be turned on at just the right time or active in precise amounts. That’s hard to do when the genes aren’t in their native habitat. Getting a gene to its normal location was no easy feat when Porteus started working on gene therapy in 1997, he says.
Then CRISPR gene editing came along. CRISPR is something like a molecular, GPS-guided pair of scissors — usually a protein called Cas9 — that seeks out and cuts DNA at precise locations. Since it debuted in 2012, researchers have been using the tool to cut, repair or insert genes in exactly the desired place in experiments with human cells grown in lab dishes and with animals (SN: 10/7/20).
Some companies have begun using CRISPR-based gene therapies in clinical trials with people. Several are taking blood-forming stem cells from patients, editing the cells in a lab, then giving the cells back to patients in a bone marrow transplant. And again, transplanting edited cells back into a patient first requires chemotherapy to kill existing bone marrow to make way for the engineered cells. But viruses aren’t always necessary to deliver the CRISPR scissors and GPS locator to the cells to be edited.
Vertex Pharmaceuticals, based in Boston, is collaborating with CRISPR Therapeutics, headquartered in Zug, Switzerland, to use CRISPR/Cas9 to break the DNA in a way that turns on fetal hemoglobin in people with either beta-thalassemia or sickle-cell disease (SN: 8/14/19). Both conditions mess with oxygen-carrying hemoglobin in red blood cells. About 60,000 people worldwide are born with symptomatic cases of beta-thalassemia each year, and sickle-cell disease is estimated to affect about 100,000 people in the United States alone, with Black and Hispanic people most likely to get the disease.
Victoria Gray, the first person to get the therapy for sickle-cell disease, in 2019, recounted her experience March 6 in London at the Third International Summit on Human Genome Editing. When her red blood cells collapsed into sickles and clogged blood vessels because of faulty hemoglobin, Gray faced sudden, unpredictable bouts of intense pain, she said. “The pain I would feel in my body was like being struck by lightning and hit by a freight train all at once.” She was often hospitalized to get blood transfusions and opioid painkillers that didn’t always help. One particularly bad episode struck in October 2010. Gray, a 37-year-old mother of four from Mississippi, said she didn’t leave the hospital until the following January. Even afterward, she couldn’t feed herself or walk on her own.
But on July 2, 2019, Gray got three vials of her own bone marrow cells edited to make fetal hemoglobin. The worst part was the recovery from chemotherapy, she said. “It took about seven to eight months for me to physically feel and mentally accept that I was better,” she told the crowd of scientists, ethicists and other experts. Thanks to her “super cells,” she now has a full-time job, attends her children’s football games and cheerleading events and can enjoy family outings. “The life that I once felt like I was only existing in, I’m now thriving in.”
The preliminary results of the clinical trials, announced in June, showed that 24 of 27 people with beta-thalassemia who previously required blood transfusions hadn’t needed a transfusion for at least a year after getting the therapy. Of 17 sickle-cell disease patients analyzed, 16 had made it at least a year without a pain crisis. All 17 avoided hospitalization for at least a year.
This approach and another sickle-cell therapy from Editas Medicine in Cambridge, Mass., break one gene to compensate for another. Porteus, of Stanford, is instead interested in using CRISPR to get a healthy replacement gene in. Such a strategy might work well for diseases where any one of thousands of mutations in a gene can be to blame, Porteus says. For cystic fibrosis, for example, 719 different mutations in the CFTR gene have been shown to cause the disease.
The team is working on techniques to increase the chances that the cuts the CRISPR scissors make will be repaired by inserting a healthy copy of the gene into the breach. This copy-and-paste method, known as homology-directed repair, rarely works, but Porteus and colleagues are making improvements. In one study, cells taken from patients with sickle-cell disease were edited and then transplanted into mice. Up to 20 percent of the defective genes had been replaced, his team reported in 2021 in Science Translational Medicine.
How CRISPR gene therapy works
Some gene therapies that may be approved soon use tools like CRISPR/Cas9 to disrupt or edit DNA directly.
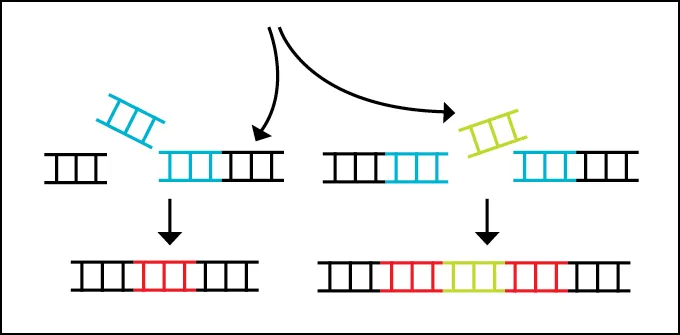
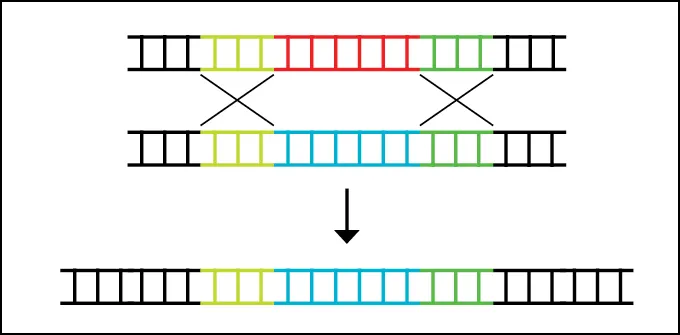
Other researchers are pursuing what’s called CRISPR base editing to alter just the typo that causes a genetic disease. This tool can fix single DNA-letter mutations, chemically changing one DNA base, the information-carrying portion of the DNA molecule (SN: 10/25/17).
Cells edited outside the body can be examined before they go back in, to ensure safety. But researchers are also trying to push CRISPR editing into the body. Those strategies, though, come with concerns over misplaced cuts that can cause stray mutations, plus some still may require potentially risky viral delivery.
In October 2022, 27-year-old Terry Hogan died after getting an AAV-delivered CRISPR gene therapy for Duchenne muscular dystrophy in a trial sponsored by the nonprofit organization Cure Rare Diseases. Hogan’s death was the result of an immune reaction to high doses of the AAV9 virus needed to carry the gene therapy to his muscles. That immune reaction set off a cytokine storm that damaged his lungs and led to his death, researchers reported September 28 in the New England Journal of Medicine.
Viral delivery isn’t the only option
Cutting, the geneticist at Johns Hopkins, has high hopes for nonviral delivery vehicles for in vivo therapies. These molecular FedEx trucks are usually composed of lipids, proteins and chemicals that form bubbles that protect the genetic cargo and transport it to where it needs to go. If you got a Pfizer or Moderna COVID-19 vaccine, you’ve already had such nanoparticles — in this case carrying messenger RNA — injected into your body. Messenger RNA, or mRNA, is an RNA copy of DNA’s instructions for building a protein.
Researchers have deployed nanoparticles to edit blood-forming stem cells in mice without removing the cells. The nanoparticles were studded with antibodies that targeted them to blood-forming stem cells in the bone marrow, says Hamideh Parhiz, a pharmacologist and biotechnologist at the University of Pennsylvania Perelman School of Medicine. The researchers also edited human cells carrying the sickle-cell mutation in the lab. In that case, the mRNA carried instructions for making a CRISPR base editor, which converted the sickle-cell mutation to a base that doesn’t cause sickling. Up to 88 percent of cells were edited, and under conditions that usually cause sickling, the crescent shapes were almost absent, Parhiz and colleagues reported July 28 in Science.
“In proof of principle, this paper is basically talking about the medicine of [the] future,” says Stefano Rivella, coauthor of the paper and a molecular biologist at Children’s Hospital of Philadelphia. He predicts such in vivo editing — because it dispenses with harvesting stem cells and chemotherapy — will be much cheaper than current gene therapies, which can cost millions of dollars. Rivella imagines vials of mRNA-loaded nanoparticles shipped to Africa or other places where there are many people with sickle-cell disease.
What does success look like?
But that’s not where we are today. Consider, for example, 14-year-old Antonio Vento. He was born with dystrophic epidermolysis bullosa, which is caused by a mutation in a gene that produces a collagen protein that helps layers of skin stay together. Antonio and others with the condition develop blisters and wounds when even slight friction tears the outer layer of their skin or mucus membranes away from the middle layer. Scarring in the mouth and esophagus can make swallowing difficult.
Extensive scarring also obscured Antonio’s vision. He had two surgeries in 2016 and 2017 on his left eye to remove the scars, but they came back two to three months later. The procedures kept failing, so “we decided not to do any additional surgeries,” ophthalmologist Alfonso Sabater of the Bascom Palmer Eye Institute at the University of Miami Health System and the Miller School of Medicine said July 24 during a news conference. There appeared to be little hope for saving Antonio’s vision.
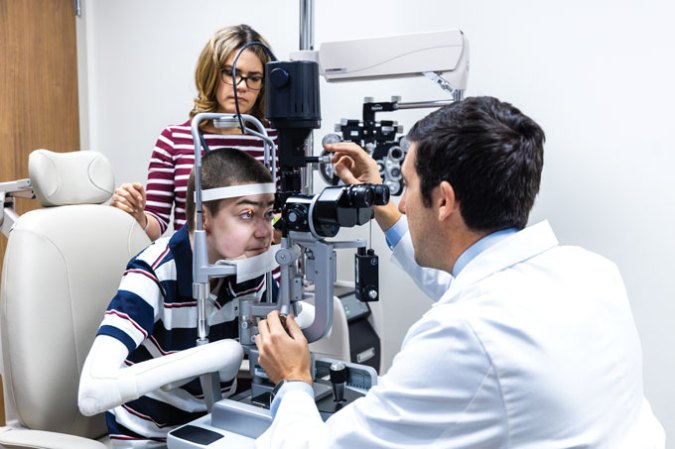
But in 2020, as part of a clinical trial, Antonio had been using a gene therapy gel developed by Krystal Biotech in Pittsburgh to keep his skin from blistering. The therapy, which is applied to the skin, uses herpes simplex virus to deliver the collagen COL7A1 gene to skin cells. It received FDA approval in May.
Desperate for a solution for his patient, Sabater worked with Krystal Biotech to reformulate the gene therapy as eye drops that might work for Antonio. The eye drops went through two years of testing for safety and efficacy. After that long haul, last year, the FDA granted permission for Antonio to get the eye drops on a compassionate use basis.
He’s the only person in the world getting the drops, which he started using weekly after surgeries to remove existing scarring. “It’s hard to say that this treatment is effective for these type of patients just with one case,” Sabater said. “But so far, the results look promising. And we’re very excited about the future, because definitely this treatment may open new possibilities.”
Antonio has recovered nearly all of his vision in one eye and about 60 percent in the other, and the scars haven’t returned. But since the effects are temporary, it’s likely that he will need regular doses for years.
Antonio’s situation highlights one of the big challenges of gene therapy. For now, it’s a boutique treatment, Porteus says, and that is expensive. The rarity of the diseases makes it hard for drug companies to recoup what they’ve invested, and thus makes them reluctant to invest to begin with.
Having the technology is not enough, notes Maria Grazia Roncarolo, the cofounder, president and head of research and development for Tr1X, a biotech company in La Jolla, Calif.
In the 1980s, Roncarolo helped pioneer a gene therapy for “bubble babies,” children with SCID caused by mutations in the gene encoding adenosine deaminase, also called ADA-SCID. While at the San Raffaele Telethon Institute for Gene Therapy in Milan, Roncarolo and colleagues reported curing two children in 2002. The pharmaceutical giant GSK licensed the therapy, called Strimvelis, and got approval from the European Medicines Agency in 2016 to use the treatment for children who don’t have a suitable bone marrow donor. “This was a beautiful story. And when the product was approved in 2016, we celebrated [with] champagne,” Roncarolo says.
Despite this approval though, patients can get the therapy only by going to Italy for compassionate use treatment. “GSK decided to disinvest in rare genetic diseases because they thought that was a market which was not big enough and profitable enough,” Roncarolo says. Orchard Therapeutics, based in London, which inherited the therapy, announced in 2020 that it was reducing its investment in ADA-SCID to focus on less rare diseases. The Telethon Institute now holds rights to the therapy and is trying to commercialize it, she says. “You can imagine the frustration for me and all the people that contributed to bring this product to the market.”
Similarly, the European Medicines Agency approved an AAV-based gene therapy for beta-thalassemia from bluebird bio in March 2019, but the company pulled the product from the European market in 2021. European health care systems were trying to negotiate lower prices for the therapy than the company thought it was worth.
Showing that a therapy is safe and effective is just one part. “The real challenge for these products,” Roncarolo says, “is to make sure — especially for rare genetic diseases — that they find a home in a company that has the motivation and resources to commercialize them.”
The stories of Antonio Vento, Will Ungerer, Victoria Gray and others show that patients are benefiting from gene therapy today. There are plenty of signs of progress over the last decades. But even if the biggest scientific hurdles fall, there remain many hurdles to fixing broken genes.