Why Huntington’s disease may take so long to develop
Repeated bits of the disease-causing gene pile up in some brain cells
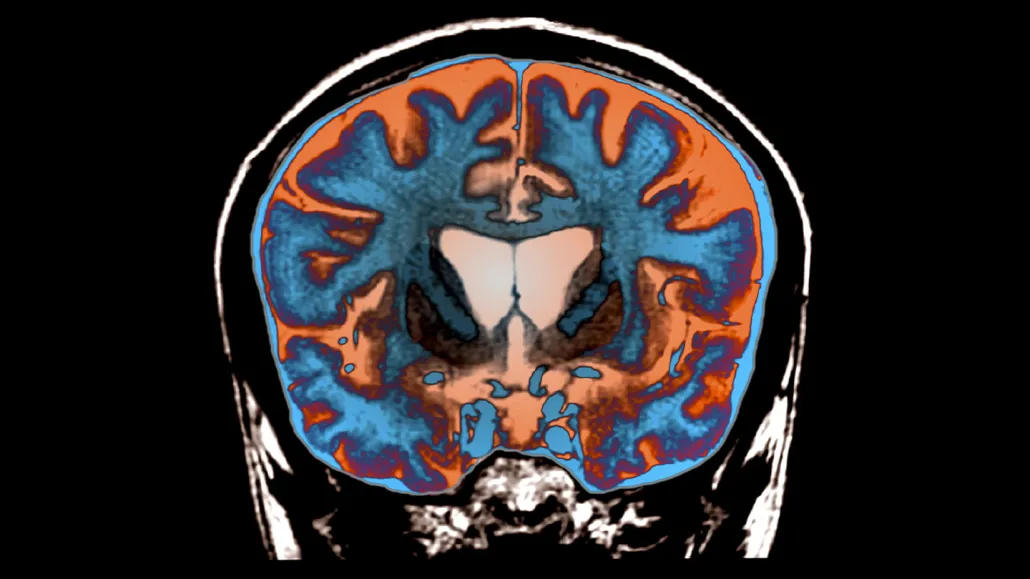
Huntington’s disease causes certain brain regions to die, as indicated by the darker areas in the center of this MRI image of the brain of a 21-year-old with the disease. Researchers have discovered that adding extra DNA to the gene that causes the disease leads to brain cell death.
Zephyr/Science Source
WASHINGTON — Scientists have uncovered a clue about why it takes so long for Huntington’s disease to develop. And they may have a lead on how to stop the fatal brain disease.
Huntington’s is caused by a mistakenly repeated bit of a gene called HTT. Until recently, researchers thought the number of repeats a person is born with doesn’t change, though repeats may expand when passed to future generations.
But in some brain cells, the repeats can grow over time to hundreds of copies, geneticist Bob Handsaker reported November 2 at the annual meeting of the American Society of Human Genetics. Once the number of repeats passes a certain point, the activity of thousands of other genes in the brain cells changes drastically, leading the cells to die.
These findings suggest that adding repeats to the HTT gene in vulnerable brain cells is what is driving Huntington’s disease, says Handsaker, of the Broad Institute of MIT and Harvard in Cambridge, Mass. The research also suggests that preventing the repeats from growing may stop the development of the disease.
The new work gives “serious insight into the disease mechanism,” says Russell Snell, a geneticist at the University of Auckland in New Zealand who was not involved in the work.
About 41,000 people in the United States have symptomatic Huntington’s disease, and another 200,000 are at risk of developing it. Inheriting just one copy of a repeat-riddled HTT gene produces symptoms.
Even though individuals are born with the disease-causing gene, symptoms don’t usually appear until people are in their 30s to 50s. Those symptoms include depression, mood swings, forgetfulness, balance problems, involuntary movements and slurred speech. Eventually, a person with the disease may be paralyzed and can die from complications such as pneumonia or heart failure.
In one part of the HTT gene, the DNA bases cytosine, adenine and guanine — CAG — are repeated over and over. Most people have 26 or fewer repeats, but people who have 40 or more repeats will develop Huntington’s disease. The more repeats, the earlier symptoms start.
Previously, researchers thought that long-term exposure to the toxic huntingtin protein — made from the disease-causing version of HTT — damaged and killed brain cells, much like smoking does to lung cells, Snell says (SN: 10/18/15).
In the new research, Handsaker and colleagues examined individual brain cells in donated brains from people with and without the disease. After measuring the length of the repeated genetic bit in all the cells, the researchers found a “dramatic expansion” of the repeats in brain cells called striatal projection neurons, also known as medium spiny neurons, in people with Huntington’s, Handsaker said. The extra DNA wasn’t found in other types of brain cells in people with or without the disease.
“Some cells had up to 1,000 CAG repeats,” Handsaker said. The additions “occurred only in the vulnerable cell types that are lost to” Huntington’s disease. Exactly why the repeats grow longer in those cells and not others is not yet known.
Only a few cells had such long stretches of repeats. Most spiny neurons had added more modest numbers, he said. Based on the ages of the donors when they died and on computer simulations, the researchers noticed an unusual pattern to the ever-accumulating repeats. It can take decades to go from about 40 repeats to 80, but then the process picks up steam. It takes only a few years to go from 90 repeats to 100s, Handsaker said.
It wasn’t clear at first whether additional repeats affected the brain cells. So, the researchers examined the activity of thousands of genes by charting RNA levels in the donated cells. The activity of those genes remained stable until the disease-causing version of HTT had about 150 repeats. Then, gene activity altered drastically: Thousands of genes increased activity, while thousands of others decreased.
Since precise levels of activity are needed to keep cells healthy, it’s probably this change that leads to the death of the cells within months of reaching 150. “It looks like a switch,” Snell says. “It looks like you’re OK, more or less, to 150.”
But, Handsaker said, why things are generally fine until that point and then go awry is another mystery. “We really don’t know what goes wrong at 150.”
Regardless, keeping the growth of the repeats in check may be a way to stop the disease from progressing. CAG sequences might inadvertently get added when a protein called MSH3, which is involved in repairing DNA, loses its place. Lowering levels of that protein may prevent the expansion, Handsaker said.
Current experimental techniques for managing Huntington’s focus on lowering the levels of huntingtin. Another insight of the new research, Snell says, is these strategies may not be necessary.
The “work represents a very interesting potential paradigm shift for [Huntington’s] research,” says Leora Fox, Assistant Director of Research and Patient Engagement for the Huntington’s Disease Society of America in New York City. “Huntingtin lowering is not dead, but targeting [DNA] instability holds therapeutic potential.”
Computational biologist Liana Lareau agrees. The work is “a success story for single-cell sequencing,” which the researchers used to examine DNA and RNA in individual cells. The new insights would not have been possible without examining how DNA changes and gene activity are coordinated, says Lareau, of the University of California, Berkeley. And brain donations were also key, she says. “We can’t see this type of thing when we look at cells … in a dish. You need the full complexity of the natural environment.”