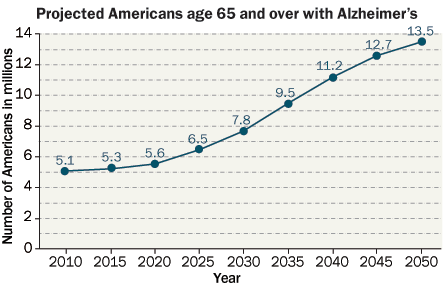
Memories Can’t Wait
Researchers rethink the role of amyloid in causing Alzheimer’s
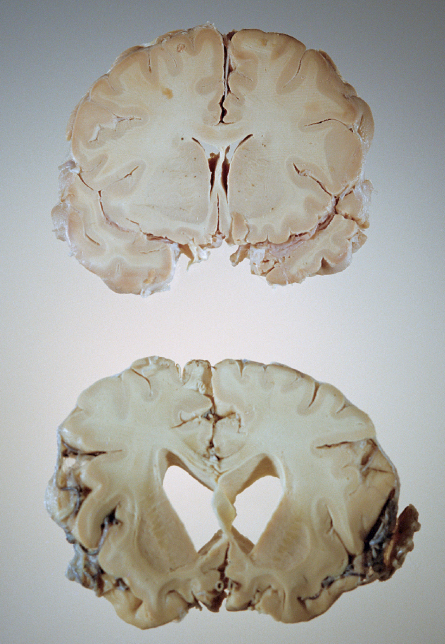
The polite term for what Alzheimer’s disease does to the brain is “neurodegeneration.”
Researchers rethink the role of amyloid in causing Alzheimer’s
The polite term for what Alzheimer’s disease does to the brain is “neurodegeneration.”