Neuron Killers
Misfolded, clumping proteins evade conviction, but they remain prime suspects in neurodegenerative diseases
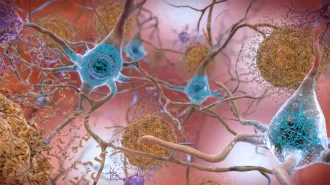
As open-and-shut cases go, Alzheimer’s disease should top the list. The victim is clear. Suspects are in custody. Wherever neurons die due to Alzheimer’s disease, a protein known as amyloid-beta is always found at the scene of the crime, hanging around in large, tough gangs called plaques. Parkinson’s and Huntington’s diseases; amyotrophic lateral sclerosis (which goes by its initials ALS or the alias Lou Gehrig’s disease); and prion diseases, such as scrapie in sheep, mad cow disease in cattle and Creutzfeldt-Jakob disease in humans, all have similar stories.
Scientific investigators have pieced together this much: A seemingly mild-mannered brain protein falls in with a bad crowd, the corrupted protein and its cronies gang up and mob violence results in the death of a brain cell. It’s a scene repeated over and over again in different neighborhoods of the brain, by different proteins, but all with the same result — the death of neurons and rise of disease.
But no one has convicted these suspected neuron killers. So far, cases mostly rely on circumstantial evidence, with large holes in the web of proof. There’s no smoking gun, no motive and no eyewitness to corroborate what scientists suspect. And there’s no cure for the diseases that slowly break down brains and spinal cords, robbing victims of memories or mobility.
No one has observed all the steps of a neuron’s demise, so no one is sure exactly what the murder weapon is or who dealt the final blow. But scientists acting like shamuses on the scent of a killer have picked up tantalizing clues about how neurons meet their end, and protein aggregation is almost certainly involved.
“It seems unlikely that coincidence is at work here,” says Bradley Hyman, a neurologist at HarvardMedicalSchool and Massachusetts General Hospital-East in Charlestown, Mass. Recent research from Hyman and colleagues shows that plaques develop more rapidly in the brains of mice prone to Alzheimer’s disease than had been thought. The discovery, published February 7 in Nature, suggests that there may be many years between the appearance of plaques and the onset of disease, providing a window of time for doctors to take action and stop the death of neurons.
Other researchers have recently reported progress on developing molecules that may help protect the brain against proteins-gone-bad. And other new research shows that the perpetrator in some cases of neurodegenerative disease may not be one of the usual suspects.
The key to stopping the killing of neurons is figuring out what causes otherwise innocuous proteins to show their Mr. Hyde side, and discovering why the proteins flock together once they’ve turned. The method by which “bad” proteins bump off neurons is also a matter of dispute. Scientists are drawing ever closer to solutions for these mysteries, and what they discover may one day help head off these diseases or even repair some damage after rogue proteins have vandalized the brain or spinal cord.
Cause or effect
Not everyone believes that protein aggregation is such a bad thing for neurons. Take those big plaques of amyloid-beta, or A-beta, found near dead and dying brain cells in Alzheimer’s disease patients.
“Some people say it’s a tombstone, others say it’s not the cause,” says Gang Yu, a neuroscientist and biochemist at the University of Texas Southwestern Medical Center at Dallas.
Big clusters of protein may be a cell’s way of coping with otherwise harmful proteins, suggests Lila Gierasch, a biophysical chemist at the University of Massachusetts Amherst. Plaques are “like garbage dumps for insoluble proteins,” she says. Indeed A-beta plaques contain remnants of other proteins, perhaps dumped in the plaque to avoid cluttering up a cell and gumming up its inner workings.
Brain images of healthy people reveal that A-beta plaques are common, even in people who don’t have dementia. And mice that make a lot of A-beta have memory problems, but their neurons don’t die, says Li-Huei Tsai, a neuroscientist at MIT. “The role of A-beta is still very, very controversial,” she says. Some people think elevated levels of the protein may interfere with neuron communication. Others think that small aggregates, rather than large clumps, are toxic to cells.
Part of the difficulty in deciphering A-beta’s role in Alzheimer’s disease is that no one is sure what the protein’s day job is. That’s true of alpha-synuclein, a protein that forms clumps called Lewy bodies inside brain cells of people with Parkinson’s disease, and of huntingtin, a protein which has been shown to be the causative agent of Huntington’s disease. Alpha-synuclein, A-beta and the prion protein PrP probably aren’t unemployed, but scientists have not yet established their roles.
On the surface, these proteins, as well as two proteins (TDP-43 and superoxide dismutase or SOD1) involved in ALS, have nothing in common, says Mark Goldberg, director of the HopeCenter for Neurological Disorders at WashingtonUniversity in St. Louis. The sequences of amino acids that compose the proteins aren’t the same, nor are the normal shapes of the proteins. The neuron-killing proteins probably function differently too. But all of them go bad in a similar way, twisting from loose, flexible molecules into rigid, sticky formations known as beta-pleated sheets.
Every protein in the body probably has the ability to form beta-pleated sheets given the right (or wrong) circumstances, says Erich Wanker, a molecular biologist and biochemist at the MaxDelbrückCenter for Molecular Medicine in Berlin. Something about these proteins and others that cause amyloidosis — fatal diseases in which abnormally folded proteins build up in organs — makes the proteins more prone to assuming the deadly conformation. Genetic mutations can tip the balance, but that doesn’t explain why people who don’t have mutations sometimes end up with the aggregates.
On the straight and narrow
Although the precipitating event that leads good proteins down the beta-pleated path isn’t known, Wanker and his colleagues may have developed a way to stop the process, at least in the test tube. In a report published in the June Nature Structural & Molecular Biology, Wanker and his collaborators showed that a small molecule called (—)-epigallocatechin gallate (mercifully shortened to EGCG) can keep A-beta and alpha-synuclein from forming beta sheets. The group had previously shown that the compound could prevent huntingtin from aggregating.
EGCG latches on to the backbones of the amino acid chains that compose the proteins. With EGCG riding piggyback, the proteins form small clumps. But apparently the proteins never switch to the beta-sheet formation, so the little clumps aren’t toxic to cells in the test tube.
Wanker doesn’t know whether EGCG, found in green tea, would be an effective therapy for neurodegenerative diseases. The researchers have yet to demonstrate that the compound can dissolve existing aggregates. Also, the experiments used equal parts of the molecule to protein in order to stop the proteins from forming the toxic beta sheets, which may mean that therapies would require massive amounts of the compound to work effectively. It’s also not known how well EGCG gets across the blood-brain barrier. If the molecule doesn’t enter the brain easily, doses of EGCG needed to prevent disease might be too high to be practical.
Cells may already possess molecules that work in the same way EGCG does, Wanker says. Proteins called chaperones also help keep other proteins loose and ready for action. Some evidence suggests that defects in chaperones may be the blow that sets off brain-wasting diseases. “This mechanism may be more common than we think,” Wanker says.
Other proteins may act as guardian angels to keep would-be neuron killers on the straight and narrow too. One such guardian may be a protein known as Pin1, which could keep another potential killer that stalks the brains of Alzheimer’s disease patients from turning deadly.
While spotlights have been trained on A-beta as the most likely killer of neurons in Alzheimer’s disease, Kun Ping Lu of BethIsraelDeaconessMedicalCenter in Boston thinks scientists may be ignoring a more deadly culprit, a protein called tau.
Tau is normally a hard-working protein that helps create the internal skeleton of the cell by binding to the cell’s frame-supporting microtubules. If not for tau, the long fibers called axons that connect neurons across the brain would break down, severing communication as surely as cutting a fiber-optic cable to a building would. Dendrites, the neuron’s branchlike projections that receive signals from other neurons, would also disintegrate without tau pinning microtubules in place.
People who have mutations in the gene that encodes tau develop a disease called frontotemporal dementia. The brains of people with this dementia look much like brains of people with Alzheimer’s disease with one critical difference: Frontotemporal dementia patients don’t have plaques in their brains. But they do have tangles of tau in brain cells, and their neurons are as dead as a person with Alzheimer’s disease.
That leads Lu to believe that tau may be more directly involved in killing neurons than A-beta. In other words, A-beta may order the hit, but tau pulls the trigger. “If, on top of tangles, you add plaques or increase A-beta, now you have massive neurodegeneration,” Lu says.
Lu lays out the scenario for brain-cell murder this way: A-beta builds up outside neurons, leading to inflammation in the brain. Inflammation prods enzymes called kinases to tack extra phosphates on to tau inside the cells. This causes tau to walk off the job and hang out in hard tangles with other tau molecules that have more phosphates hanging off them than groupies on a rock star. Hyperphosphorylated tau forms such tight bonds with its cronies, not even boiling it in detergent can untangle it, Lu says. After that, it’s all over for the neuron as its axons and dendrites collapse.
Normally, tau’s protector, Pin1, keeps it from falling in with hardened tangles. Pin1 actually does double duty, watching over tau and APP, the protein precursor to A-beta. Mutations in the gene for Pin1 have now been linked to late-onset Alzheimer’s disease, but not to early onset forms.
Lu and his colleagues have found a variation in the Pin1 promoter, a stretch of DNA that controls activity of the gene, associated with a five-year later onset of Alzheimer’s disease. The researchers don’t yet know if the variation increases Pin1 production. They do know that aging causes Pin1 production to fall.
“As people get older and older, Pin1 levels drop, drop, drop,” Lu says.
Boosting Pin1 levels may help untangle tau in people at risk of Alzheimer’s disease, slowing the disease’s progression or preventing it altogether. Reporting in the May Journal of Clinical Investigation, Lu and colleagues showed that making more of the protein could help protect against tangle formation in mice. But the new research also shows that too much Pin1 can be a bad thing. When researchers increased Pin1 levels in mice carrying the P301L alteration in tau — found in people with frontotemporal dementia — more brain cells died than did in mice that carry the tau mutation but make normal levels of Pin1.
The poisoning blame game
Tau is not the only protein that may be getting away with neuron murder while a more high-profile suspect takes the rap. The antioxidant protein superoxide dismutase had been fingered as the killer of spinal cord neurons in people with ALS. A small subset of those with the disease have mutations in the gene for SOD1 that lead to clumping of the protein and the death of neurons that direct motion.
But recently scientists learned that nearly everyone with ALS has aggregates of a protein called TDP-43 (for TAR DNA binding protein) in their spinal neurons.
“If TDP-43 is the major pathway, then SOD1 was misdirecting us,” says Christopher Shaw, a neurologist and neurogeneticist at King’s College London. He estimates that about 1 percent of people with ALS have mutations in the gene for TDP-43. Shaw and his colleagues showed in a report published March 21 in Science that those mutations lead the protein to stick together more readily. Most cases are sporadic, not inherited, and occur when TDP-43 twists into a shape that favors aggregation. Scientists don’t yet know what sets off the conversion, but Shaw says the tail of the molecule certainly plays a role.
The tail end of TDP-43, what scientists refer to as the c-terminus, “aggregates fantastically quickly,” he says. “It’s an extremely sticky little beast.”
That stickiness is characteristic of all proteins that form neuron-killing beta sheets and may account for the speed at which plaques and other aggregates form. Although scientists have evidence that proteins become toxic after twisting into beta sheets and aggregating, just how clumps of protein poison neurons isn’t clear.
For instance, even though SOD1 protein is made everywhere in the body, and mutations that lead to overproduction cause aggregation of the protein in many tissues, only spinal cord neurons degenerate to give rise to ALS. Similarly, neurons that produce dopamine, a chemical key to neural communication, are the victims of alpha-synuclein clumps in people with Parkinson’s disease. And Alzheimer’s plaques tend to congregate in parts of the brain that are active when people are daydreaming or thinking about nothing in particular .
The life cycle of a neuron might explain its susceptibility to damage, Shaw says. Most neurons last a lifetime. The cells don’t divide after they are born and take their place in the brain. Some new neurons do develop in parts of the brain, but most of the 10 billion to 100 billion neurons are present before birth and last until death. The cells never get a day off and they have no backup or replacement.
Their long lives may lead neurons to produce proteins differently than other cells. “Maybe brain cells have a just-in-time policy,” Shaw says. “You don’t make a lot of protein and stack it up, so therefore you don’t have the same rigorous protein turnover mechanisms.” In other cells in the body, quality control would quickly recognize a misfolded protein and get rid of it before it could cause mischief. The lack of supervision in neurons could make them more vulnerable to rogue proteins.
On the other hand, neurons may process proteins correctly, but age may catch up with the neurons, making them weary of the constant effort against aggregation.
“There’s an ongoing battle for many years, and ultimately the neuron gives up,” speculates Yu from UT Southwestern. But scientists don’t know what causes neurons to throw in the towel. The final straw could be the loss of chaperone proteins, which oversee protein-folding, or a strike by the cellular machinery that transports or breaks down proteins, causing crowding in the cell that foments aggregation.
“Theories abound,” Yu says, “but none have been definitively proven.”