Bacterial soft rot can severely damage a wide variety of fruits and vegetables. Infected potatoes, for example, develop mushy areas that render them inedible.
The chief culprit is a bacterium called Erwinia chrysanthemi. It attacks a plant by secreting various proteins, including an enzyme known as pectate lyase C. This microbial enzyme initiates soft rot by cleaving a major molecular component of a plant’s cell walls. Now, mathematicians have come up with a computer program to help identify bacteria with this and other disease-causing enzymes.
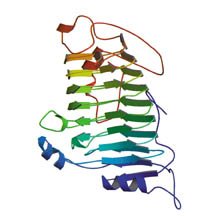
Like all proteins, pectate lyase C consists of a long string of amino acids with its own characteristic sequence. The string is intricately folded into a compact glob. Unlike many other proteins, however, this enzyme folds itself into a pattern of parallel segments that coil into a large helix. This protein configuration, or structural motif, is called a beta helix.
When Frances A. Jurnak, now at the University of California, Irvine, and her coworkers first determined the enzyme’s three-dimensional structure in 1993, they speculated that its unusual folding pattern might be found in other proteins that incorporate similar sequences of amino acids.
Since then, researchers have determined the structures of several proteins that also have beta-helix folds. One example is pertactin, a protein made by Bordetella pertussis, the bacterium responsible for whooping cough.
Applied mathematician Bonnie A. Berger of the Massachusetts Institute of Technology (MIT) and her collaborators have developed a novel computer program that sorts through amino acid sequences of proteins with unknown structures to pick out those likely to contain a beta helix.
When they used the program, the researchers were surprised to discover that their top candidates for containing beta helices were all bacterial proteins. “We found [evidence of] beta helices in many bacterial proteins, but not in human or mice proteins,” Berger says. “The beta helix may be the mechanism by which bacteria infect their hosts.”
The list of beta-helix candidates includes proteins called virulence factors, which promote the entry of bacteria into host cells; adhesins, which bind to cell surfaces; and toxins produced by the intestinal bacterium Helicobacter pylori, which causes gastrointestinal disease.
Berger described her group’s findings at the Joint Mathematics Meetings, held in January in New Orleans. The researchers’ results are expected to guide biologists in selecting proteins for structural analysis.
Protein sequences
Organisms manufacture thousands of different proteins, which act as enzymes, structural elements, or carriers. Molecular biologists have developed laboratory techniques for rapidly determining a protein sequence, and databases include hundreds of thousands of these sequences. The sequences constitute only part of the protein story, however. Each protein strand folds itself into a particular three-dimensional shape to perform its biological function.
Determining a protein’s shape is a slow, painstaking process that can take a laboratory several months. Using X-ray crystallography and other methods, researchers have so far worked out the structures of roughly 15,000 proteins. “There are many more known sequences than there are known structures,” Berger notes.
Because a protein’s shape is the key to understanding its biological function and because protein-folding mistakes have been implicated in ailments such as Alzheimer’s disease, researchers have been trying to accelerate the structure-determination process.
“Given the long and painful nature of these experiments, it would be extremely valuable to have good computational approaches to predict the structure of a protein based on its sequence,” Berger contends. Such predictions can guide biologists in choosing which proteins to analyze further.
From a computational biology standpoint, “the protein-folding problem is very difficult, partly because a given protein may fold into different configurations under different conditions,” she says. Furthermore, various seemingly unrelated amino acid sequences can fold into the same configuration.
In practice, researchers often tackle a simpler task than that of predicting a protein’s folded structure based on its amino acid sequence. They concentrate on determining whether a sequence encodes a particular folding pattern, or motif.
Strings of amino acids generally arrange themselves into one of two patterns. In an alpha helix, amino acids coil into a spiral, with 3.6 amino acids making up each turn of the helix. In a beta sheet, a string of amino acids folds accordion-fashion into parallel segments oriented in alternating directions to form a flat sheet.
A less common, more recently identified structure is the beta helix. There, parallel strands that would normally lie in a flat sheet instead curl into a helix. Each turn of a beta helix consists of three beta segments, with 22 amino acids per turn, so the structure is about five times as wide as an alpha helix.
“We believe that the beta helix is a primitive or simple beta sheet,” says MIT biologist Jonathan King.
A given protein may contain one or more instances of each of these features, along with some loops, links, and other miscellaneous folds.
Coiled-coil structures
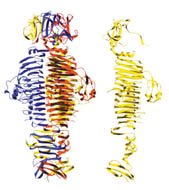
Helices sometimes participate in more complex structures. Two alpha helices–from adjacent parts or separate parts of the same amino acid string–can wrap around each other into a so-called coiled-coil structure. Initially, Berger and her colleagues focused their research on computational methods for identifying strands within protein sequences that would form coiled coils.
“We focused on coiled coils because they are one of the most interesting and important motifs from a biological point of view,” Berger says. Coiled coils are often found in membrane-fusion proteins, which play a role in permitting the human immunodeficiency virus (HIV) and other viruses to enter a cell. They also have been found in proteins that control cell growth.
To search for coiled coils, Berger’s team adopted a statistical approach that checks how often each of the 20 different types of amino acid arises in a particular protein. Amino acids in coiled-coil structures repeat in a cyclic pattern of seven positions along a string, which amounts to about two turns of the helix. In some of these seven positions, certain amino acids tend to occur more frequently than others. For example, the amino acid leucine is roughly three times as likely to occur in the first position of a seven-unit segment as in any other particular position along the segment. Moreover, some amino acids never appear in certain positions.
In 1995, Berger and MIT colleague David B. Wilson developed the PairCoil computer program to identify two-strand coiled coils by looking for correlations between pairs of amino acids. With this statistical analysis, the program assigns a score to each position in a sequence. A high score means that a particular amino acid is likely to be in a coiled-coil structure.
To include three-strand coiled coils, the PairCoil motif-recognition program evolved into the more versatile MultiCoil version. Additional tweaks, including the adoption of a learning algorithm, led to LearnCoil versions. These can identify coiled-coil motifs in the proteins that viruses secrete and enable them to enter cells.
When they applied the LearnCoil program to databases of sequences, the researchers found evidence of coiled coils in fusion proteins associated with a wide variety of disease-causing viruses, including the influenza virus, Moloney murine leukemia virus, HIV, simian immunodeficiency virus, and visna virus.
Guided by the LearnCoil results, Peter S. Kim, who was at MIT but is now at the Merck Research Laboratories in West Point, Pa., and his collaborators, synthesized and analyzed proteins of the visna virus. “The structures were exactly as predicted,” Berger says. “With a good procedure to find candidate coiled coils quickly and accurately, time spent in the lab can be minimized.”
Kim and his group have used PairCoil and its successors to study coiled-coil structures and elucidate the mechanisms by which viruses invade cells. The programs have been very successful at distinguishing coiled coils from other sequences, the researchers say.
Tailspike model
Other scientists are focusing on the beta helix. King has a particular interest in this structure because it is found in a protein produced by a bacteria-infecting virus that he has long studied. “We have used the protein as a model for the study of how amino acid sequences determine protein folding and misfolding,” King says.
Known as the P22 tailspike, this protein binds with long, stringy, sugar-based structures that stick up from the surface of Salmonella bacteria. In effect, the protein permits the virus to infect its host, King says.
He and his coworkers have been meeting regularly with Berger and her group of mathematicians. Several years ago, for example, the two teams developed a novel model of how a virus protein folds itself into the proper shape to form part of a virus’ outer shell (SN: 3/25/95, p. 186).
Berger’s team took up the challenge of predicting which lengths of amino acids within P22 tailspike would fold into beta helices. Identifying a beta-helix structure in a protein sequence proved trickier, however, than identifying a coiled-coil structure.
One problem is that amino acids that are close together in the three-dimensional helical arrangement are far apart in the protein’s sequence. Another is that researchers have only a dozen known structures to use as examples against which to test prediction techniques.
Berger, Phil Bradley of MIT, and Lenore J. Cowen of Johns Hopkins University in Baltimore developed an algorithm that identifies sequences that have a high probability of folding into a beta-helix structure. The result was a new computer program called BetaWrap.
The BetaWrap program searches for a landmark amino-acid sequence to find a potential starting point for a beta helix. It then assigns scores to nearby amino-acid positions and sums corresponding scores from rung to rung of the putative helix. As in previous motif-recognition programs, a high score represents a high probability that the beta-helix structure is present.
When Berger’s group tested the BetaWrap program on protein sequences with known structures, it readily picked out all the beta-helix proteins. Let loose on a large protein-sequence database, it generated a lengthy list of candidates. Interestingly, the complete list includes allergens, such as ragweed pollen. In 1993, Jurnak and her collaborators had suggested, based on sequence similarities, that plant-pollen proteins might also contain beta-helix structures.
Several candidates in Berger’s list had higher scores than the known beta-helix proteins, and the top 100 were all proteins produced by bacteria. This suggests that beta-helix proteins, which feature spiky, protruding loops, may be intimately involved in attaching to or penetrating cell membranes.
The elongated shape of the beta helix makes it particularly suited to binding the floppy sugars that cover the surfaces of many cells. “Binding to such molecules may be a way for many pathogenic microorganisms to recognize and invade host cells,” King suggests.
Bradley reported the research teams’ results last month in Montreal at the computational molecular biology meeting RECOMB 2001.
Understanding structure
Now, biologists have a strong incentive to use X-ray crystallography and other imaging techniques to determine the structures of the proteins on Berger’s list and to take a closer look at how they function. “If you understand the structure, you might be able to get a better handle on how to stop an infection,” Berger says.
“If one can really identify unknown sequences that are important in bacterial pathogenesis, that could be very useful in developing the next generation of antimicrobials and diagnostics,” King comments.
Before that can happen, however, researchers need to confirm BetaWrap’s accuracy by determining the actual structure of some of the proteins on the BetaWrap list of candidates. They will consider the algorithm successful only if those proteins actually prove to contain beta-helix structures.