Unsolved drugs
A simmering controversy surrounds what’s still unknown about how antibiotics kill
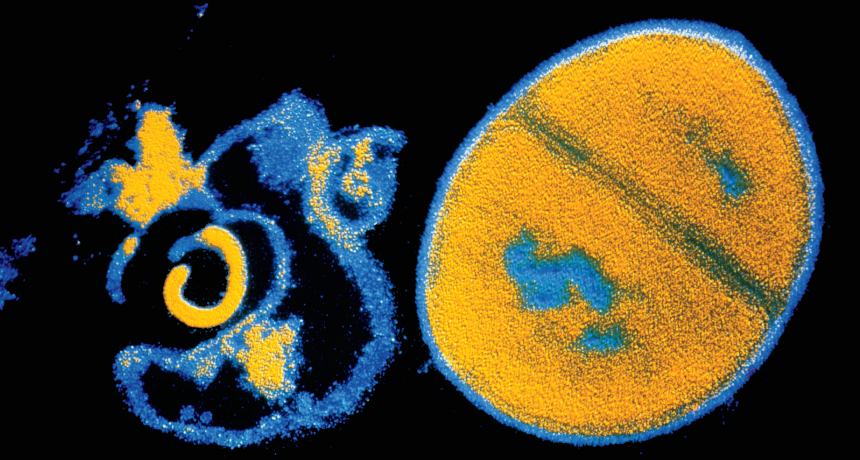
BATTLING BACK Antibiotics such as penicillin attack a bacterium’s cell wall, bursting it (left, shown next to an intact Staphylococcus aureus cell in false color). Some studies suggest that antibiotics also trigger other kinds of damage that could be harnessed in the battle against antibiotic resistance.
CNRI/Science Source
Penicillin attacks with a calculated strike, splitting open cell walls.