The first CRISPR therapy approved in the U.S. will treat sickle cell disease
The gene-editing treatment aims to help people with recurring pain crises
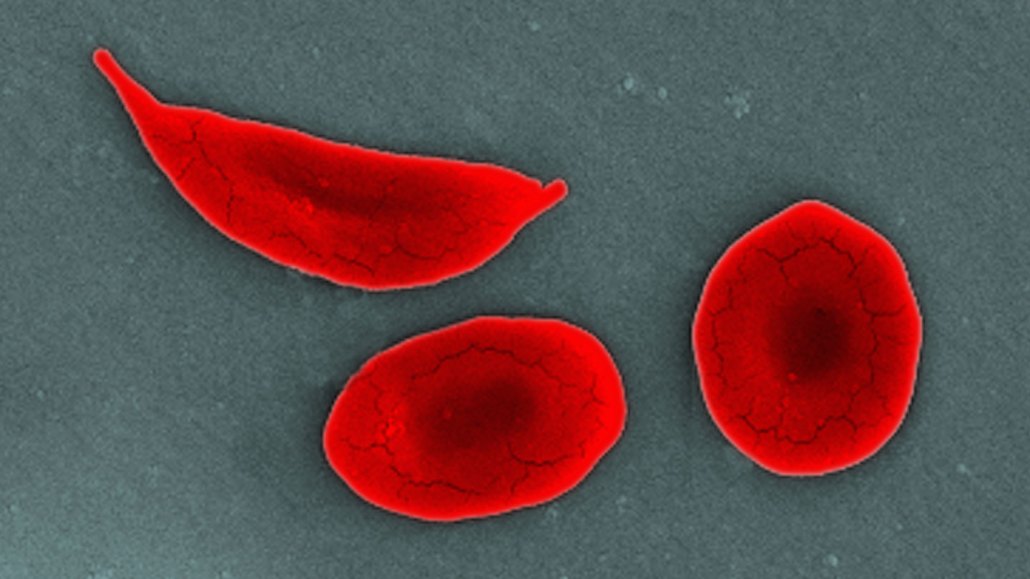
People with sickle cell disease have inflexible, curved blood cells (shown in this SEM image) that get stuck in blood vessels and block blood flow to tissues, causing bouts of debilitating pain.
Callista Images/Getty Images